Table of Contents
Medical Device registration in Europe or EU (Entire CE Marking process)
Please click here the European commission medical device website and for more information on medical device registration in Europe.
1) The first thing you need is to get a qualified PRRC (person responsible for regulatory compliance). Usually it is the same person who the management representative as defined by ISO 13485. However this can be sub-contracted to a qualified consultant (such as CMS).
2) Next you need to classify your device in accordance to Annex VIII of Medical Device Regulation (MDR). You can get the latest copy from the European commission website link above. Please treat the information from this link as the source of all original information. It is NOT recommended to find the latest CE regulation changes from any consultant website (including ours!). You can also get guidance on classification of products from MEDDEV documents (even though it might be more application to the old MDD).
3) Developed a Quality Management System (QMS) according to ISO 13485. If you are class I self-certified product, there is no need to get audited by a Certification Body (Such as TUV, BSI, SGS etc). The reason why you want to developed a QMS first before doing up technical documentation is because you can start documenting your specifications, design inputs, outputs in an orderly and official manner. This is to prevent the need to do retrospective documentation after the design of your product is out.
4) Developed the technical file according to Annex II and III of MDR. You can visit here to see how can do the General Safety and Performance Requirements. The format is slightly different from the Essential Requirement checklist from the old MDD. Visit here for layman explanation on the requirement for technical fie. Visit here to see the mistakes to avoid when drafting the clinical evaluation report (CE). Please note CER is not that simple to pass CE requirements anymore under MDR and Meddev 2.7.1 Rev 4. Even experience regulatory personnel may need to seek experienced clinical writers to review or draft the CER for the manufacturer. Click here to see other mistakes to avoid when transiting from MDD to MDR.
Also remember to get your Unique Device Identifier (UDI) from organization like GS1. See here to see UDI implementation timeline. Also you would need to select your conformity assessment route. Most companies would choose Annex IX as the default. Again this is not needed for Class I self certified product.
5) Draft the EU DOC (Declaration of Conformity) before the external audit. This is needed because the EC REP or Notified Body usually wants to see how you draft the EC DOC. Click here to see an example.
6) Appoint an EC REP if the manufacturer does not reside in EU.
7) Get audited by Notified Body. If you are class I self-certified product, there is no need to get audited by a Notified Body (Such as TUV, BSI, SGS etc). Usually for the first time, manufacturers get ISO 13485 certificate before getting CE certification. However some manufacturer prefer to get both at the same time. For annual surveillance audit, manufacturers prefer to use one service provider to do the external audit to prevent multiple audit in one year.
8) So now you have the UDI, CE Cert and EC REP. You can now ask your EC REP to do medical device registration in Europe (where the EC REP reside) via EUDAMED! After this step you can now launch your product in EU and claim that your Medical Device has CE Marking!
Now there are some flow charts that may ask you to engage your EC REP earlier or draft the EU DOC later. But these are not important so long you ensure all 8 steps are done up completely.
Click here to contact us on more information or on our services for medical device registration in Europe.
Medical Device Registration in Europe: Actor Module coming in 1 Dec 2020 (section updated on 26 Oct 2020)
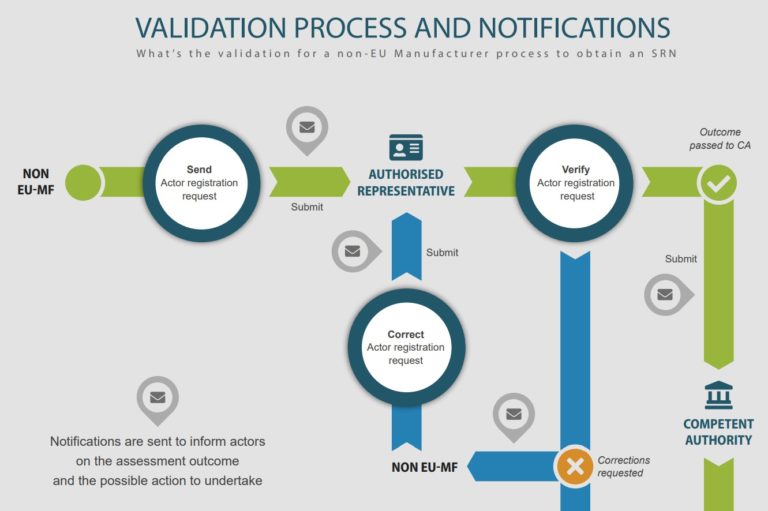
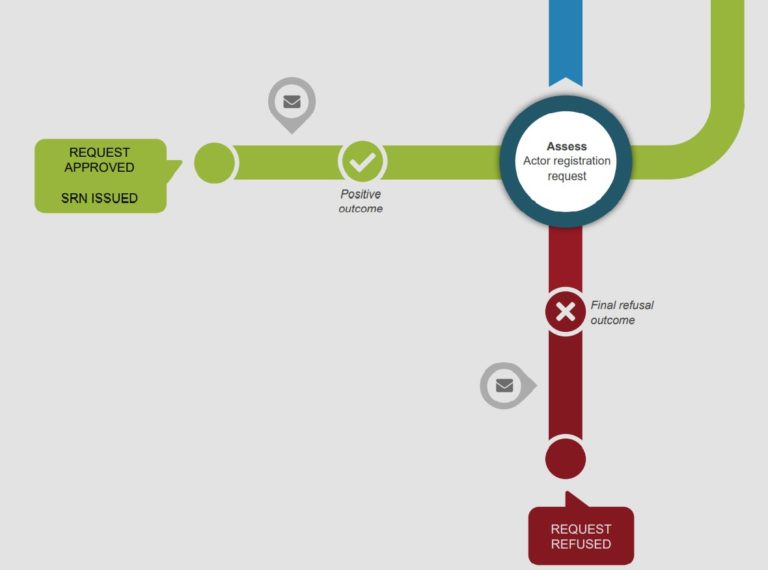
Coming 1st December 2020, the European Commission will make the Actor registration module available. This would be the first of six EUDAMED modules that would launched eventually.
Economic operators would be able to submit information through the Actor registration module to obtain a single registration number (SRN) when it is launched. With the SRN, economic operators can be clearly identified publicly. Please note that economic operator refer to the following: EU and non-EU manufacturers, authorised representatives, system/procedure pack producers and importers.
Documents to provide with the actor registration request:
1 Declaration on information security responsibilities
All actors must upload a signed Declaration on information security responsibilities
2. Mandate Summary document
To register in EUDAMED, the non-EU manufacturers must have an active authorised representative and submit with the registration a Mandate Summary document
For more information, please visit here. Please see infographic (from EC) below for an illustration of the process.