Therapeutic Goods Administration (TGA) is responsible for evaluating, assessing and monitoring medical devices to help Australians stay healthy and safe.
Medical Device Definition
Medical devices include a wide range of products, such as medical gloves, bandages, syringes, blood pressure monitors, and x-ray equipment. They differ from medicines as they generally have a physical or mechanical effect on the body or are used to measure (or monitor) the body and its functions. While the objective of these products is to help improve your health and wellbeing, it’s important to know that their use also has potential risks.
Medical devices are defined in the Therapeutic Goods Act 1989 as any of the following items for human use:
instrument
apparatus
appliance
software
implant
reagent
material or other article.
Medical devices are used to:
diagnose, prevent, monitor, predict outcomes of, treat, or ease symptoms of medical conditions
replace or enhance parts of the body
control or support conception
examine specimens from the human body.
These products play an important role in health care, so it’s important they’re safe to use and work properly.
In vitro diagnostic (IVDs) are typically tests that are used to examine human samples to assist with diagnosing and managing a patient’s health. IVDs are regulated as medical devices. They include reagents, kits, calibrators, controls, laboratory analysers and software, as well as tests used at the point of care, or in the home, such as COVID-19 rapid antigen self-tests and glucose monitoring tests by diabetics.
Medical Device Classification
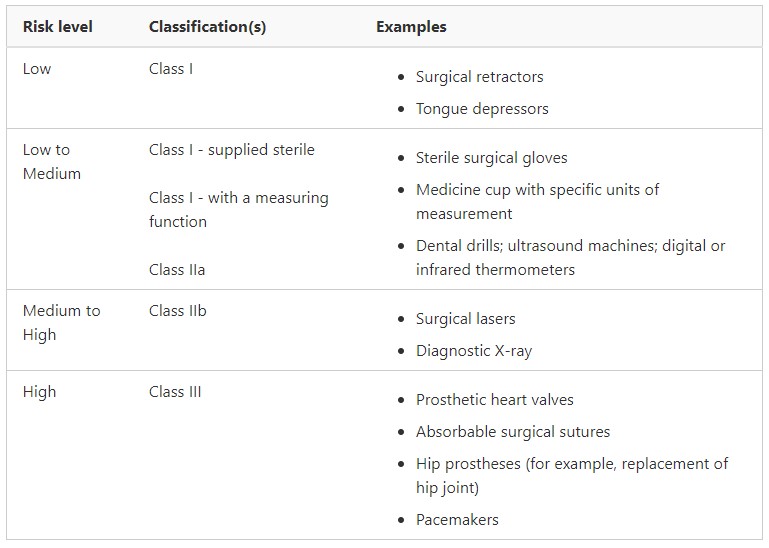
Medical Device Classification (non IVD)
Medical devices will be placed into one of the main classifications depending on the level of risk they pose. The higher classification level, the tougher the requirements will be.
Devices are classified by considering a number of different questions, such as:
What does the manufacturer intend the medical device to be used for?
How invasive will it be in the body (e.g. is it a bandage to be placed on the skin, or a catheter to be inserted into the body)?
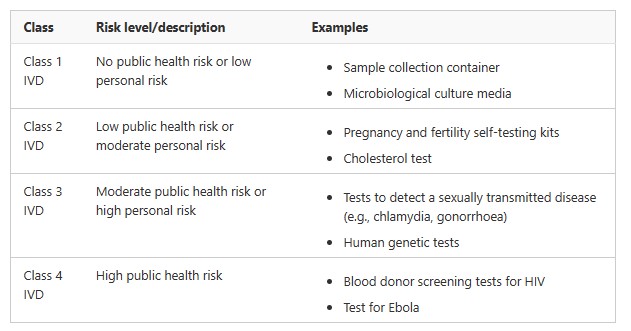
The classification of IVD medical devices is based on their intended purpose and the public health risk or personal risk that may arise from an incorrect result.
The higher the potential risk an incorrect result would pose, the higher the classification.
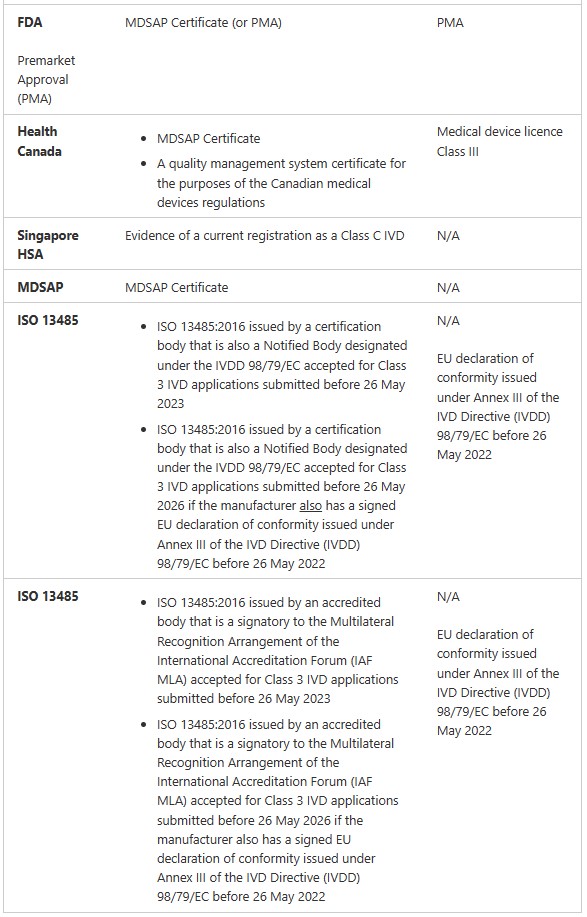
Examples of IVD medical devices with different classifications are summarised in the following table.
More information about the classification of IVD medical devices can be found in Schedule 2A of the MD Regulations and Classification of IVD medical devices.
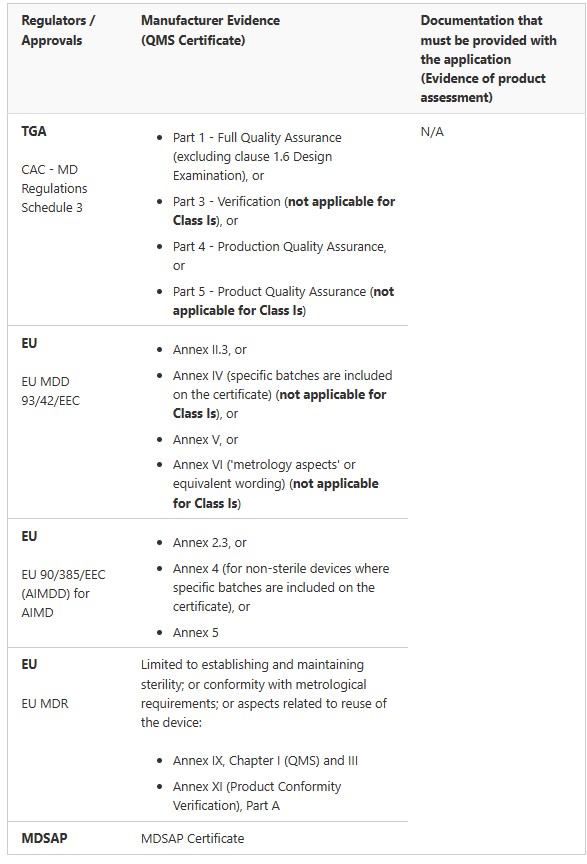
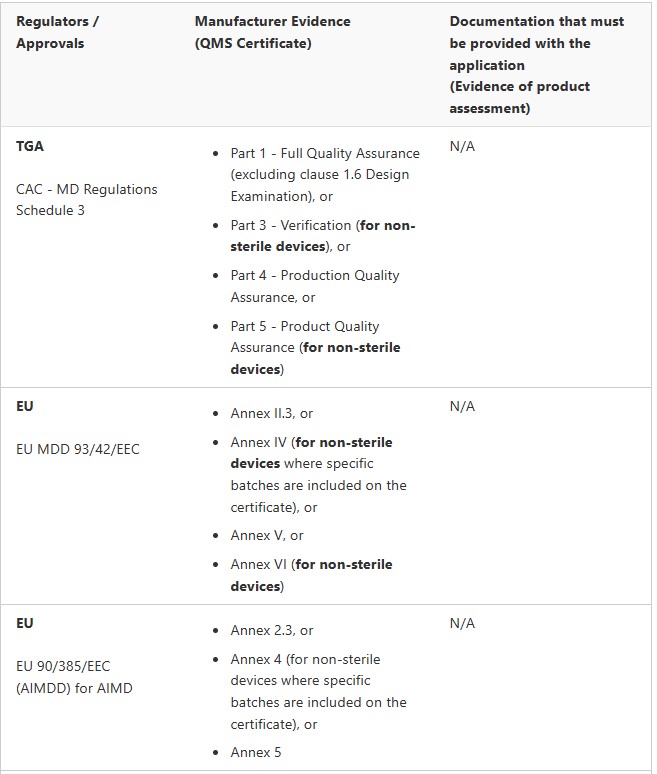
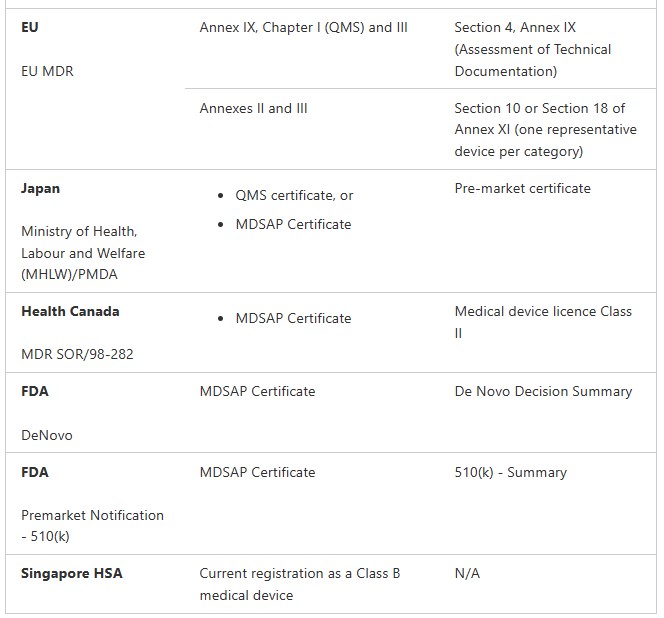
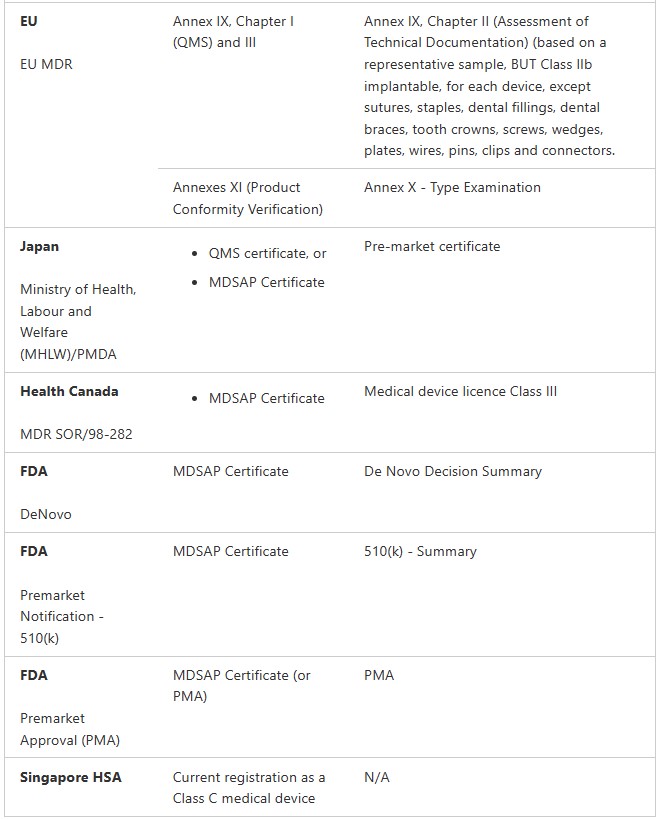
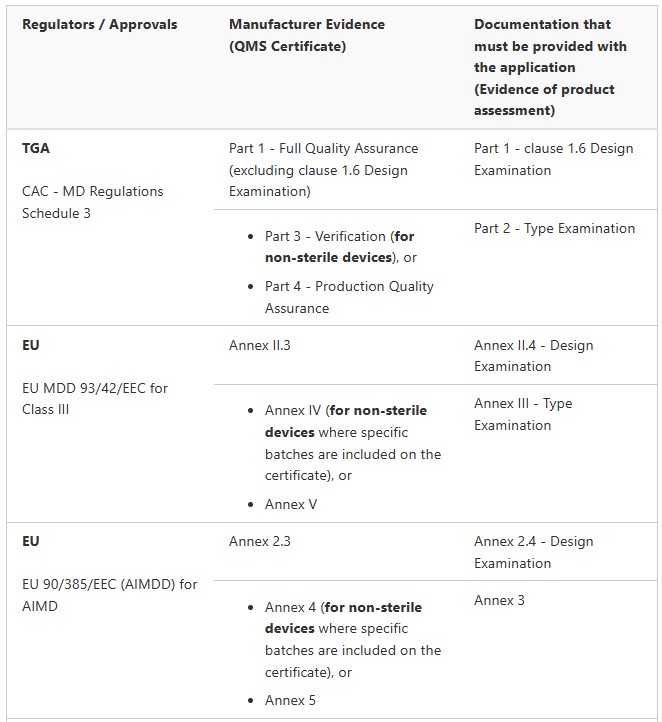
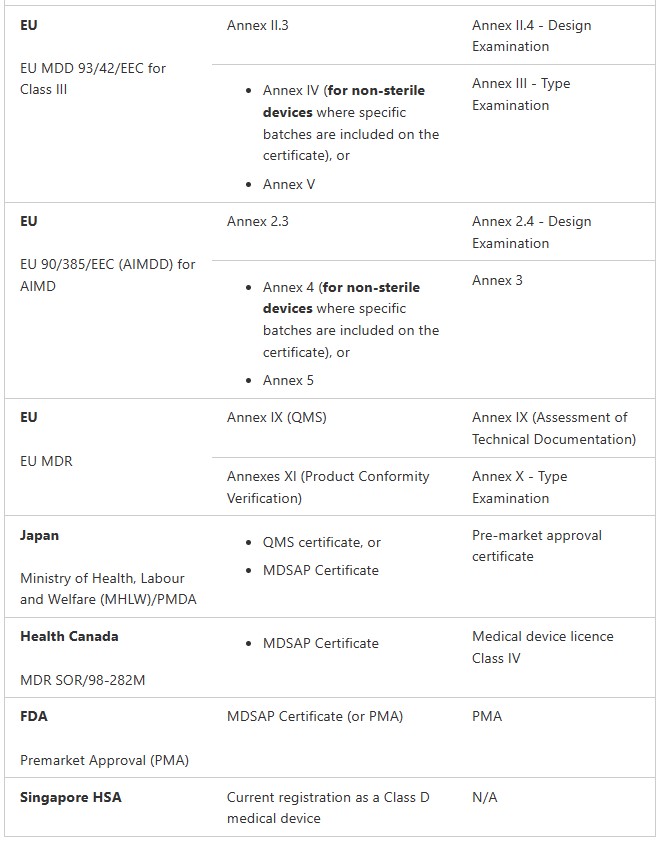
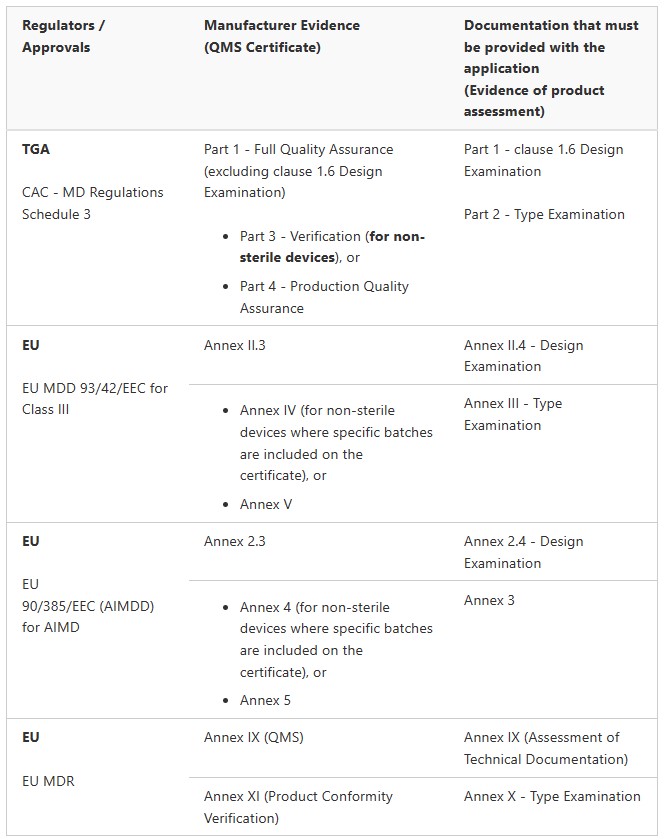
Medical Device Registration Pathways: TGA Conformity Assessment
Medical Devices (not including IVDs)
Class I non-sterile, non-measuring (Regulation 3.9(1))
Class Is or Im (supplied in a sterile state or with measuring function) (Regulation 3.9(2) and 3.9(3))
Class IIa (Regulation 3.8)
Class IIb (Regulation 3.7)
Class III and AIMD (Regulation 3.6 (except specified medical devices). These are medical devices that do not contain medicines or materials of animal, microbial, recombinant or human origin. Class III and AIMD (Regulation 3.6)
Class III and AIMD (Regulation 3.6) (specified medical devices) These are medical devices contain medicines or materials of animal, microbial, recombinant or human origin.
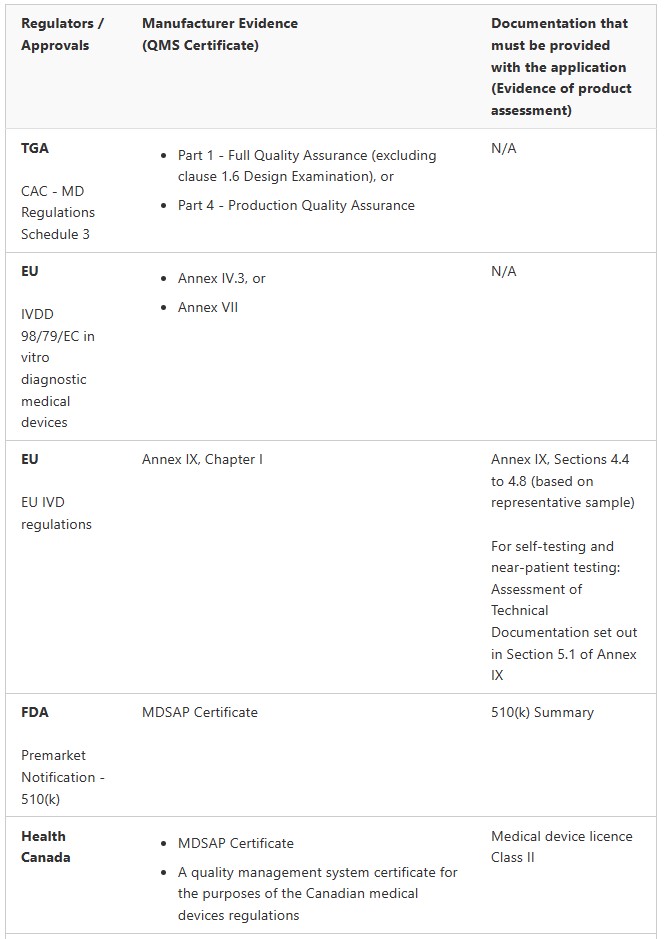
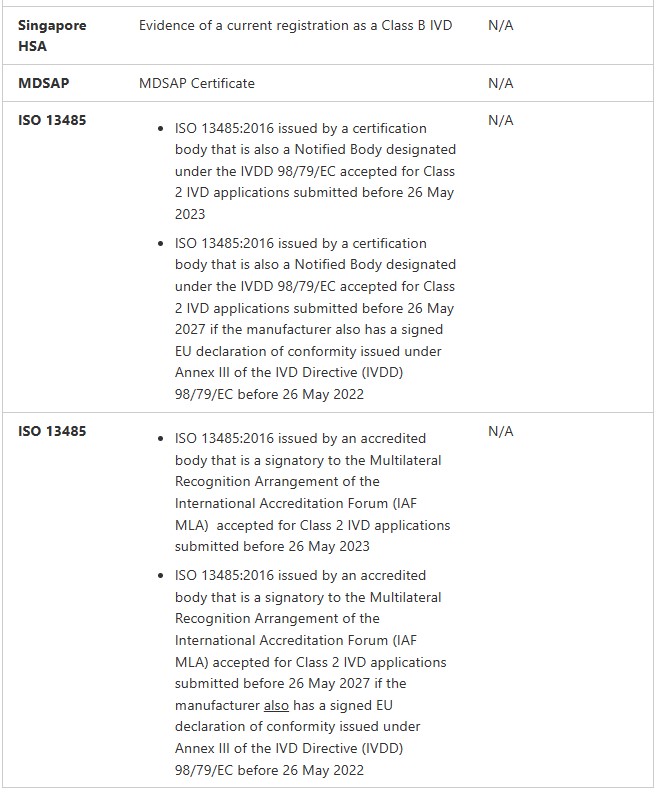
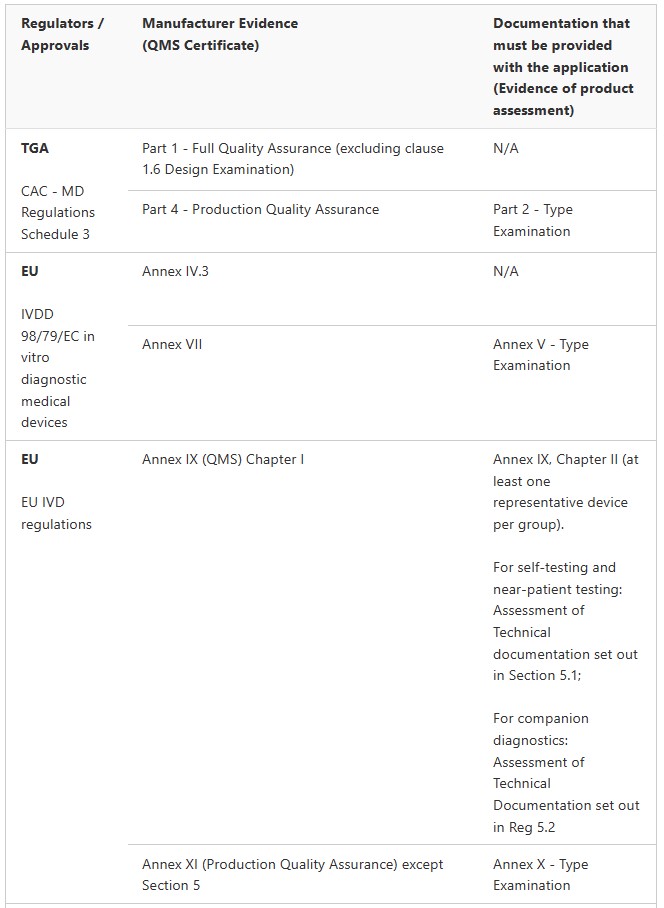
IVDs
Class 1 IVD and Class 1 IVD Export only (Regulation 3.9A)
Class 2 IVD (Regulation 3.8A)
Class 3 IVD (Regulation 3.7A)
Class 4 IVD (Regulation 3.6A)
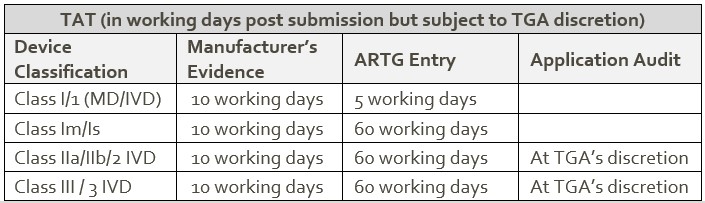
Application audits
Application audits are conducted to verify that devices submitted for inclusion in the ARTG meet the relevant legislative requirements.
For some applications, an audit is mandatory under the legislation. Others may be selected for auditing at the discretion of the delegate.
For more information whether your device requires application audit, please visit this link: https://www.tga.gov.au/resources/resource/guidance/auditing-medical-device-including-ivd-medical-device-applications
Document Requirements
• DOC -TGA • Essential Principles (EP) • Applicable Device information- Intended purpose & Indication • Risk Management Report (plan, analysis and report) • FMEA Report • Labelling & Instruction for Use • Post market surveillance plan • Vigilance System • Manufacturing Information and Design • Usability Engineering File • Clinical Evaluation Report (CER) • Biocompatibility Report (Cytotoxicity, Irritation, Sensitization) • Biological Evaluation Report • Sterilization validation (if applicable) • Cleaning validation • Safety and performance testing • Software development plan including software risk assessment (if device contains software) • Transport validation • Service Life Report • Stability testing EC / ISO Certificate (if applicable)
Quality Management System
Two key requirements of the conformity assessment procedures for medical devices are that a manufacturer must implement:
a Quality Management System (QMS) for the design, production, packaging, labelling and final inspection of a device, and
inspection and quality assurance techniques that are to be applied during the production of a device
The Therapeutic Goods (Conformity Assessment Standard for Quality Management Systems) Order 2019– external site specifies relevant medical device standards for quality management systems and medical devices intended to be supplied in a sterile state. The Order ensures that if a manufacturer’s QMS, or inspection and quality assurance techniques, comply with the relevant standards specified in the Order, the TGA will treat the QMS, or the inspection and quality assurance techniques, as if they comply with the parts of the conformity assessment procedures specified in the Order.
Apart from ISO13485:2016 for QMS, this order also identifies a number of other ISO standards, or parts of ISO standards, referred to in the former Order that have been updated by ISO since that order was first introduced. For example:
ISO 11137-2:2006 Sterilization of health care products Radiation Part 2: Establishing the sterilization dose, has been revised by ISO 11137-2:2013 Sterilization of health care products Radiation Part 2: Establishing the sterilization dose; and
ISO 13408-1:1998 Aseptic processing of health care products Part 1: General requirements, has been revised by ISO 13408-1:2008 Aseptic processing of health care products Part 1: General requirements.
ISO standards are often adopted by regional or national standards organisations. Where a regionally or nationally adopted version of an ISO standard has been applied and the adopted version of the standard is identical to the original parent publication of the ISO standard, the TGA treats these standards as being interchangeable with the original parent ISO standard.
For example, the standard ISO 13485:2016 – Medical Devices – Quality management Systems- Requirements for regulatory purposes has been adopted as an identical regional or national standard in several jurisdictions:
EN ISO 13485:2016 (also I.S. EN ISO, DIN EN ISO, BS EN ISO, NSAI ISO etc.) in Europe,
ANSI/AAMI/ISO 13485:2016 in the USA, and
AS ISO13485:2017 in Australia.
While the Order only specifies the parent ISO 13485:2016 standard, where a manufacturer has met the requirements of an identical regional/national adopted standard the TGA considers that the requirements of ISO 13485:2016 have also been met.
License Transfer: Changing the sponsor of therapeutic goods
The ‘sponsor’ of therapeutic goods here refers to the person or company in relation to whom goods are registered, listed, or included in an entry in the Australian Register of Therapeutic Goods (ARTG).
Changes to the sponsorship of therapeutic goods in an entry in the ARTG occur where:
the sponsor dies, is made bankrupt, or (if they are a company) the business is wound up, or
the sponsor transfers or assigns its therapeutic goods business or their interest in inclusion of the goods in the Register to the person to whom the business or interest is transferred or assigned (the new sponsor).
As a result of such events, the new sponsor becomes responsible for the therapeutic goods in the ARTG entry, and must notify us of that event.
You, as the sponsor, are also required to notify us if you change your name, or amalgamates with another company under another name.




 COVID-19 Medical Device Registration in Malaysia
COVID-19 Medical Device Registration in Malaysia
