



Table of Contents
Introduction: Easy Guide on how to comply to MDR and ISO 13485
“Easy Guide on how to comply to MDR and ISO 13485” is aimed to provide first the essential list of procedures that needs to be in place to comply to ISO 13485. Next it would introduce elements that needs to added to this quality management as well as adding and indicating what CE technical documentations consist of and where it can be retrieved from your ISO 13485: 2016 Quality Management System (QMS) to comply with MDR. Yes this is the all-in-one guide that would attempt to present a practical checklist for you to ensure you have all the essential elements in your documentations to comply to CE MDR and ISO 13485: 2016!
Please also note that this checklist is a live document and we have added MDSAP elements to this checklist. Lastly we would upload free templates as they become available!
How to use “Easy Guide on how to comply to MDR and ISO 13485”
“Easy Guide on how to comply to MDR and ISO 13485” would be organise according to the ISO 13485: 2016 numbering system (because we got to admit it is a good numbering system!). Next there would be advise how and where are the possible locations whereby you can add requirements from CE MDR to your QMS. Please get a copy of CE MDR from here.
Easy Guide on how to comply to MDR and ISO 13485 contents
4.1 General requirements & 4.2.1 other documentation specified by applicable regulatory requirements.(document)

There could be a process for issue of EU declaration of conformity DOC). This should include the identity of the person/ title who has the authority to sign it. The person most likely to sign on it would be the Person Responsible for Regulatory Compliance (PRRC) or Management Representative (MR) who could be the QA/ RA director/ manager or even a consultant. There should also be instructions to ensure that the DOC is available in languages required by the Member State(s) in which the device are made available. MDR reference: Annex IX, 2.1
 Mandate (and letter of intention) from EU Authorized Representative (if manufacturer is not resided in the EU). This is typically known as the EC REP agreement previously. MDR reference: Annex IX, 2.2 (b) 4th
Mandate (and letter of intention) from EU Authorized Representative (if manufacturer is not resided in the EU). This is typically known as the EC REP agreement previously. MDR reference: Annex IX, 2.2 (b) 4th
 Manufacturer need to get a Single Registration Number (SRN) for registration of medical device in EU. MDR reference: Article 31
Manufacturer need to get a Single Registration Number (SRN) for registration of medical device in EU. MDR reference: Article 31
 UDI Registration and Process. MDR reference: Article 10(7)
UDI Registration and Process. MDR reference: Article 10(7)

Update of Harmonized Standards and CS process. This should be concluded in Management Review. MDR reference: Article 10(9)
 Translations for label MDR reference: Article 10(11), for implant card MDR reference: Article 18 and EU DOC MDR reference: Article 19(1)
Translations for label MDR reference: Article 10(11), for implant card MDR reference: Article 18 and EU DOC MDR reference: Article 19(1)
 Significant changes to be reported to Notified Body as indicated in MDR reference: Annex IX 2.4. Please ask you Notified Body for a change form if they have one.
Significant changes to be reported to Notified Body as indicated in MDR reference: Annex IX 2.4. Please ask you Notified Body for a change form if they have one.
 Summary of Safety and Clinical Performance for implantable and Class III as per MDR reference: Article 32
Summary of Safety and Clinical Performance for implantable and Class III as per MDR reference: Article 32
 Systems and procedure pack requirements as per MDR reference: Article 22
Systems and procedure pack requirements as per MDR reference: Article 22
 Technical Documentation as per MDR reference: Annex II and III
Technical Documentation as per MDR reference: Annex II and III
- EU DOC with UDI-DI
- Risk Management Plan
- Risk Management File
- Risk Management Report
- Clinical Evaluation Plan
- Clinical Evaluation Report
- Clinical Investigation Documents
- Device Description and Specification
- Labels and Instruction for Use
- Design information (can be reference to QMS)
- Manufacturing process and validations
- Identification of sites (including OEM)
- Results of preclinical testing
- PMS Plan
- PMS Report or PSUR
- PMCF
United States (FDA):
Confirm that the medical device organization has determined the applicability of unique device identifier requirements per 21 CFR 801 and 21 CFR 830, has obtained the unique device identifiers from an FDA-accredited UDI-issuing agency, and the required data elements have been entered in the Global Unique Device Identification Database (GUDID) [21 CFR 801, 830].
4.1.1 Role of Organization (document)
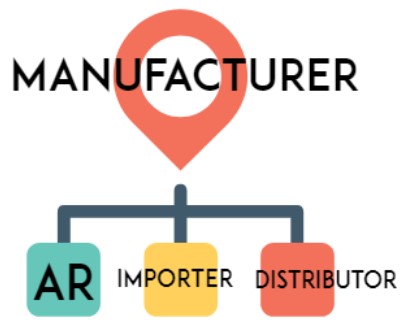
Australia (TGA):
Manufacturer of a medical device is the person who is responsible for the design, production, packaging and labeling of the device before it is supplied under the person’s name, whether or not it is the person, or another person acting on the person’s behalf, who carries out those operations. A manufacturer of a medical device is also the person who, with a view to supplying the device under a person’s name, does one or more of the following using ready made products: assembles, packages, processes, refurbishes, labels the device, or assigns a different intended purpose through the use of labels, instructions for use, advertising, or technical documentation (TG Act s41BG).
Australian importers (Sponsors) are required to include (register) medical devices from non-Australian Manufacturers in the Australian Register of Therapeutic Goods (ARTG). Sponsors are required to register the Manufacturers that they represent and to obtain a Client ID and Location ID for the manufacturer from the TGA.
To assist the Australian Sponsor, Manufacturers, who are supplying product to the Australian market and choose to participate in the MDSAP, must undertake the following to demonstrate that they have met the obligations on Manufacturers who wish to supply to Australia;
– Classify the device using the Australian classification rules
– Identify from the Therapeutic Goods (Medical Devices) Regulation 2002, an Australian conformity assessment procedure that is to be applied in accordance with the classification of the device
– Select a relevant GMDN term
– Obtain an MDSAP audit of their QMS and their device technical documentation in accordance with this Audit Approach, for demonstrating that the QMS requirements of the selected conformity assessment procedure have been applied.
– Prepare an Australian Declaration of Conformity in accordance with the requirements of the Conformity Assessment Procedure that has been applied [TG(MD)R Sch 3 P1 Cl1.8].
– Enter into a written agreement with the Sponsor. See Annex 4 for guidance on the roles and responsibilities of the Australian Sponsor and an Overseas Manufacturer
Note: If the manufacturer chooses to participate in the MDSAP for any reason, and product is supplied to the Australian market, the requirements for QMS in a relevant conformity assessment procedure must be included with the scope of the audit performed by a recognized MDSAP auditing organization.
Note: Sponsors are required to provide the Manufacturer with information in relation to the Manufacturer’s obligations under a conformity assessment procedure and information in relation to whether the devices comply with the Essential Principles [TG Act 41FN(3)(e)].
Refer to following:
Therapeutic Goods Act 1989
– Part 4-2 – Essential Principles and medical device standards
– Part 4-3 – Conformity Assessment Procedures
– Part 4-5 – Including medical devices in the Register
– Part 4-9 – Public Notification and recall of medical devices
– Chapter 5 – Advertising
Therapeutic Goods (Medical Devices) Regulations 2002
– Part 5 – Division 5.2 – Conditions
– Schedule 1 – Essential Principles
– Schedule 2 – Classification Rules
– Schedule 3 – Conformity Assessment Procedures
Brazil (ANVISA):
Manufacturer means any person who designs, manufactures, assembles or processes finished devices, including those who only perform sterilization process, labeling and packaging [RDC ANVISA 16/2013: 1.2.9].
For a domestic manufacturer, confirm that the establishment has ANVISA’s authorization to manufacture medical devices (AFE – Autorização de Funcionamento da Empresa). For domestic and international manufacturers, verify that the products already distributed in the Brazilian market are registered/notified with ANVISA [Brazilian Federal Law nº 6360/76].
According Brazilian Legislation, the Good Manufacturing Practice (GMP) certification is a prerequisite for medical device registration. Therefore, the facility site inspection precedes the device registration request. Medical devices subject to notification do not need the GMP certificate, but even not being certified, their manufacturers shall comply with the GMP requirements.
Medical devices registration/notification
Device marketing authorization shall be requested to ANVISA by the domestic manufacturer or importer (legal representative) formally established in Brazil. Registration is a comprehensive process for market authorization, applied to medical devices in classes III and IV. [ANVISA RDC nº 36/2015, RDC nº 40/2015]
Notification is a simplified market authorization process, applied to all medical device classes I and II. [ANVISA RDC nº 36/2015, RDC nº 40/2015]. Registration is valid for 10 years, while notification has no expiry date. Renewal of the registration shall be requested upon time defined at Brazilian Law 6360/1976.
Establishment license
Domestic manufacturer: shall be authorized by ANVISA, at a minimum, as a manufacturer of medical devices. This license includes authorization to store and distribute medical devices.
Importer: the importer is considered the legal representative of the international manufacturer in Brazil and shall be authorized by ANVISA to import, store, and distribute medical devices. In the case of outsourcing the storage, the importer does not need authorization for this activity.
Canada (HC):
Manufacturer means a person who sells a medical device under their own name, or under a trade-mark, design, trade name or other name or mark owned or controlled by the person, and who is responsible for designing, manufacturing, assembling, processing, labeling, packaging, refurbishing or modifying the device, or for assigning to it a purpose, whether those tasks are performed by that person or on their behalf [CMDR 1].
No person shall import or sell a Class II, III or IV medical device unless the manufacturer of the device holds a license in respect of that device or, if the medical device has been subjected to a change described in section 34, an amended medical device license [CMDR 26].
An application for a medical device license shall be submitted to the Minister by the manufacturer of the medical device in a format established by the Minister [CMDR 32].
An application for a medical device license shall include a copy of a quality management system certificate certifying that the quality management system under which the medical device is manufactured (class II) or designed and manufacturer (class III or IV) satisfies National Standard of Canada CAN/CSA-ISO 13485:2016. [CMDR 32(2)(f); 32(3)(j); 32(4)(p)].
Japan (MHLW):
“Marketing Authorization Holder” means a person who resides in Japan and is granted a license for marketing from a prefectural government [PMD Act 23-2.1].
Application or Notification for marketing
Class 2, class 3, and class 4 medical devices except for the ones specified by the requirement of PMD Act 23-2-23.1.
An” Application for Marketing Approval” shall be submitted to PMDA by the Marketing Authorization Holder to get authorization for marketing a medical device in Japan. [PMD Act 23-2-5.1]
An “Application for QMS Audit” shall also be submitted to PMDA by the Marketing Authorization Holder, when they do not have an effective QMS Certificate for the device. [PMD Act 23-2-5.6, 7]
Class 2 and class 3 medical devices which are specified by the requirement of PMD Act 23-2-23.1
An” Application for Marketing Certification” shall be submitted to a Registered Certification Body (RCB) by the Marketing Authorization Holder to get authorization for marketing a medical device in Japan. [PMD Act 23-2-23.1].
An “Application for QMS Audit” shall also be submitted to an RCB by the person, when the person does not have a valid QMS Certificate for the device. [PMD Act 23-2-23.3, 4].
A “Notification for Marketing” shall be submitted to PMDA by the Marketing Authorization Holder for marketing a class 1 device in Japan [PMD Act 23-2-12].
A class 1 medical device doesn’t need any QMS Certificate for marketing.
Facility Registration (Registered Manufacturing Site)
A medical device manufacturing site which conducts one of the designated manufacturing processes listed below shall be registered:
– Main Designing
– Main assembly
– Sterilization
– Domestic storage before final release.
The site is called “Registered Manufacturing Site”. It has to submit an application to PMDA for registration by itself [PMD Act 23-2-3.1, 23-2-4].
United States (FDA):
21 CFR 807 – Establishment Registration and Device Listing for Manufacturers and Initial Importers of Devices.
Establishment means a place of business under one management at one general physical location at which a device is manufactured, assembled, or otherwise processed.
Owner or operator means the corporation, subsidiary, affiliated company, partnership, or proprietor directly responsible for the activities of the registering establishment.
Owner or operator must register the establishment and submit listing information to Food and Drug Administration (FDA) for those devices in commercial distribution, regardless of classification.
The registration and listing requirements must pertain to any person who:
– Initiates or develops specifications for a device that is to be manufactured by a second party for commercial distribution by the person initiating specifications
– Manufactures for commercial distribution a device either for itself or for another person; regardless of whether the manufacturer places the device into commercial distribution or returns the device to the customer
– Repackages or relabels a device
– Acts as an initial importer, except that initial importers may fulfill their listing obligation for any device for which they did not initiate or develop the specifications for the device or repackage or relabel the device by submitting the name and address of the manufacturer – Manufactures components or accessories which are ready to be used for any intended health-related purpose and are packaged or labeled for commercial distribution for such purpose
– Sterilizes or otherwise makes a device for or on behalf of a specification developer or any other person
– Acts as a complaint file establishment
– Is a device establishment located in a foreign trade zone.
Australia (TGA):
Marketing authorization (inclusion in the Australian Register of Therapeutic Goods [ARTG]) is granted to the Australian Sponsor. The Sponsor cannot apply for marketing authorization until the Manufacturer has completed a conformity assessment procedure that is relevant for the Class of the device. Non-Australian Manufacturers will need to assist the Sponsor through the provision of information to support an application for marketing authorization and to meet the relevant conditions for on-going supply. A Sponsor is provided with a Certificate of Inclusion in the ARTG to identify the products that have been granted marketing authorization. Products with marketing authorization may be identified from the public facing ARTG database.
A Sponsor is normally not permitted to import, supply, export, or manufacture (in Australia) a medical device unless; the device complies with the essential principles, the Manufacturer has applied a relevant conformity assessment procedure, and the Sponsor has included the device in the ARTG. An exemption to allow importation and supply may be approved and granted to the Sponsor by the TGA, in cases where the TGA is satisfied that the device is to be used under a clinical trial scheme or the special access or authorized prescriber schemes. An exemption approval may contain conditions in relation to the manufacture of the product. The Manufacturer should verify with the Australian Sponsor if any such conditions have been applied to the exemption approval.
A Manufacturer must maintain a list of their Australian Sponsors and the products those Sponsors have included in the Australian Register of Therapeutic Goods.
A Manufacturer’s procedures should ensure that product is not released for supply to the Australian market unless the Sponsor has been issued with a “Certificate of Inclusion in the Australian Register of Therapeutic Goods”, that identifies each kind of medical device that has been approved for supply to the Australian market [TG Act s41FJ], or the Sponsor holds a relevant exemption (TG Act Part 4-7).
As part of an application for marketing authorization a Sponsor commits to certain requirements that are identified in s41FD – “Matters to be certified”. These matters include establishing a written agreement with the manufacturer about the provision of information and establishing effective communication channels for post-market activities. See Annex 4 for further guidance.
Brazil (ANVISA):
In Brazil there are two kinds of marketing clearance, registration and notification:
– Device market clearance shall be requested to ANVISA by the domestic manufacturer or importer (legal representative) formally established in Brazil. Registration is a comprehensive process for market authorization, applied to medical devices in classes III and IV. [ANVISA RDC nº 36/2015, RDC nº 40/2015].
– Notification is a simplified market authorization process, applied to all medical devices classes I and II. [ANVISA RDC nº 36/2015, RDC nº 40/2015] Registration is valid for 10 years, while notifications have no expiry date – renewal of the registration shall be requested upon time defined at Brazilian Law 6360/1976.
Canada (HC):
No person shall import or sell a Class II, III or IV medical device unless the Manufacturer of the device holds a license in respect of that device or, if the medical device has been subjected to a change described in section 34 – an amended medical device license [CMDR 26].
Japan (MHLW):
Any person who intends to market a medical device for business in Japan shall have a license for marketing granted by the prefectural government. This person is called a “Marketing Authorization Holder” (MAH) and shall reside in Japan [PMD Act 23-2.1]. The person has to submit an Application for Marketing Approval/Certification (class 2, 3 or 4 medical device) or a Notification for Marketing (class 1 medical device) to get marketing clearance for the medical device. No person shall market a medical device in Japan, unless the Marketing Authorization Holder of the device has been granted the marketing clearance [PMD Act 23-2-5.1, 23-2-23.1, 23-2-12].
United States (FDA):
21 CFR 807.81- Premarket Notification:
Each person who is required to register his establishment pursuant to 807.20 must submit a premarket notification submission to the Food and Drug Administration at least 90 days before he proposes to begin the introduction or delivery for introduction into interstate commerce for commercial distribution of a device intended for human use which meets any of the following criteria:
– The device is being introduced into commercial distribution for the first time; that is, the device is not of the same type as, or is not substantially equivalent to, (i) a device in commercial distribution before May 28, 1976, or (ii) a device introduced for commercial distribution after May 28, 1976, that has subsequently been reclassified into class I or II.
– The device is being introduced into commercial distribution for the first time by a person required to register.
21 CFR 814 – Premarket Approval
A Premarket approval is required for any FDA class III device that was not on the market (introduced or delivered for introduction into commerce for commercial distribution) before May 28, 1976, and is not substantially equivalent to a device on the market before May 28, 1976, or to a device first marketed on, or after that date, which has been classified into class I or class II.
Australia (TGA):
The Manufacturer is required to notify their auditing organization body of:
– A proposed change to their QMS, including the name of location of the manufacturer
– A proposed change to critical suppliers or the goods and services they provide
– A proposed change to a validated manufacturing process
– A proposed change to the kinds of medical devices to which the system is to be applied
– For Class III or AIMD, a proposed change to the design, intended performance, intended user, packaging, storage or transport conditions of a device.
Changes are to be evaluated by the Auditing Organization to determine whether a special audit is required to verify the continuing integrity of the quality management system, or whether verification of the change may occur at the next routine audit. The Auditing Organization should also verify the continuing adequacy of technical documentation as a result of the change (see Annex 1)
If the Manufacturer is a holder of a TGA Conformity Assessment Certificate, then the Manufacturer is also required to notify the TGA of these changes, prior to implementation. For changes that are not considered substantial by the Manufacturer or applicant, they should be notified to the TGA at the time of recertification of an existing conformity assessment certificate, included within the scope of another conformity assessment application, or made available for the auditor during the next on site audit; whichever occurs earlier.
Examples of substantial changes that may require notification to the TGA include, but are not limited to, the following:
– Name and/or address of the Manufacturer
– Scope of existing manufacturing facilities, including manufacturing steps
– Addition or removal of a manufacturing facility along with associated activities
– Critical manufacturing process (e.g. a drug coating process, a sterilization method etc.)
– Critical supplier and/or relevant scope
– Type of conformity assessment procedure
– Device category – Product design (e.g. materials for medical devices, storage, shelf-life, and packaging)
– Information to be provided with a medical device (e.g. intended purpose of the device in the IFU, removal of warnings, contraindications, or other information regarding safety etc.)
Refer to:
Therapeutic Goods (Medical Devices) Regulations 2002
– Regulation 3.5 – Medical devices manufactured outside Australia
– Schedule 3 – The relevant conformity assessment procedure chosen by the Manufacturer
Note: An entry in the Australian Register of Therapeutic Goods (inclusion) in the name of the Australian Sponsor is in effect until cancelled.
Brazil (ANVISA):
Changes involving medical devices already approved by ANVISA, shall be submitted for a new approval [Brazilian Law nº 6360/76 – Art. 13]. Changes/modifications that shall be submitted are those ones classified as significant change, which affects:
– features of safety and effectiveness, including measures to communicate information (ex. residual risk)
– identification of the device or its manufacturer or manufacturing site
– indication for use, including its purpose, patient type (adult, pediatric, newborn)or environment to be used (domestic, hospital, ambulance, etc.)
– device classification
– technical specification of the device, including composition and other operational/technical/physical features
– manufacturing method.
Examples of modifications that may require a submission include, but are not limited to, the following:
– Sterilization method
– Structural material / composition
– New or additional manufacturer
– Manufacturing method
– Manufacturing site
– Operating parameters or conditions for use
– Patient or user safety features
– Sterile barrier packaging material
– Stability or expiration claims
– Design – Labels and instructions of use (if modification is regarding information)
– Commercial name
– Indication for use
– New software version
– Commercial presentation
– Inclusion of a new device in a family of medical devices already approved
– Inclusion of new accessories.
Canada (HC):
If the Manufacturer proposes to make one or more changes, the Manufacturer shall submit to the Minister, in a format established by the Minister, an application for a medical device license amendment including the information and documents set out in section 32 that are relevant to the change [CMDR 34].
Every Manufacturer of a licensed medical device shall, annually before November 1 and in a form authorized by the Minister, furnish the Minister with a statement signed by the Manufacturer or by a person authorized to sign on the Manufacturer’s behalf describing any change to the information and documents supplied by the Manufacturer with respect to the device, other than those to be submitted under section 34 or 43.1 [CMDR 43].
If the holder of a medical device license discontinues the sale of the medical device in Canada, the licensee shall inform the Minister within 30 days after the discontinuance, and the license shall be cancelled at the time that the Minister is informed [CMDR 43(3)].
Subject to section 34, if a new or modified quality management system certificate is issued in respect of a licensed medical device, the Manufacturer of the device shall submit a copy of the certificate to the Minister within 30 days after it is issued [CMDR 43.1].
Japan (MHLW):
A change to a medical device which is approved/certified by PMDA/a Registered Certification Body may require the Marketing Authorization Holder to submit a new application, a change application, or a change notification [PMD Act 23-2-5.1, 23-2-5.11, 23-2-5.12, 23-2-23.1, 23-2-23.6, 23-2-23.7].
Changes that require the application or the notification are those ones which directly impact the safety and efficacy of the device and/or the substantial identity of the fact approved during marketing approval / certification.
The Registered Manufacturing Site shall communicate with the Marketing Authorization Holder about the change when the Registered Manufacturing Site plans such changes, so that the Marketing Authorization Holder could take any necessary regulatory actions mentioned above [MHLW MO169; 29].
Examples of changes that may require an application or a notification include, but are not limited to, the following:
– Design
– Composition
– Raw material
– Sterilization method
– Manufacturing method
– Manufacturing site
– Patient or user safety features
– Operating Parameters or conditions for use
– Indication for use
– Shelf life
– Performance Specification.
United States (FDA):
21 CFR 807 – Establishment Registration and Device Listing for Manufacturers and Initial Importers of Devices.
Update the device listing information during each June and December or, at its discretion, at the time the change occurs. Conditions that require updating and information to be submitted for each of these updates are as follows:
– If an owner or operator introduces into commercial distribution a device identified with a classification name not currently listed by the owner or operator
– If an owner or operator discontinues commercial distribution of all devices in the same device class
Update registration if changes in individual ownership, corporate or partnership structure, or location of at the time of annual registration, or by letter if the changes occur at other times. This information must be submitted within 30 days of such changes. Changes in the names of officers and/or directors of the corporation(s) must be filed with the establishment’s official correspondent and must be provided to the Food and Drug Administration upon receipt of a written request for this information.
21 CFR 807.81- Premarket Notification:
A new complete 510(k) application is required for changes or modifications to an existing device, where the modifications could significantly affect the safety or effectiveness of the device, or the device is to be marketed for a new or different indication. All changes in indications for use require the submission of a 510(k).
Examples of modifications that may require a 510(k) submission include, but are not limited to, the following:
– Sterilization method
– Structural material
– Manufacturing method
– Operating parameters or conditions for use
– Patient or user safety features
– Sterile barrier packaging material
– Stability or expiration claims
– Design.
21 CFR 814.39 – PMA Supplements
After FDA’s approval of a PMA, an applicant shall submit a PMA supplement for review and approval by FDA before making a change affecting the safety or effectiveness of the device for which the applicant has an approved PMA. While the burden for determining whether a supplement is required is primarily on the PMA holder, changes for which an applicant shall submit a PMA supplement include, but are not limited to, the following types of changes if they affect the safety or effectiveness of the device:
– New indications for use of the device
– Labeling changes
– The use of a different facility or establishment to manufacture, process, or package the device
– Changes in sterilization procedures
– Changes in packaging
– Changes in the performance or design specifications, circuits, components, ingredients, principle of operation, or physical layout of the device
– Extension of the expiration date of the device based on data obtained under a new or revised stability or sterility testing protocol that has not been approved by FDA
– An applicant may make a change in a device after FDA’s approval of a PMA for the device without submitting a PMA supplement if the change does not affect the device’s safety or effectiveness and the change is reported to FDA in post approval periodic reports required as a condition to approval of the device, e.g., an editorial change in labeling which does not affect the safety or effectiveness of the device.
Australia (TGA):
Confirm that the Manufacturer’s procedure for dealing with substantial changes to a critical process (e.g. sterilization, processing materials of animal origin, processing materials of microbial or recombinant origin, or processes that incorporate a medicinal substance in a medical device), requires the Manufacturer to notify the Auditing Organization of their plans before implementing a change to a critical process. The Auditing Organization is to assess the proposed change before implementation by the Manufacturer, to determine if the requirements of the relevant conformity assessment procedure will still be met after the change. [TG(MD)R Sch3 P1 1.5(2)].
If the Manufacturer is also a holder of a TGA Conformity Assessment Certificate, then the Manufacturer is also required to notify the TGA of these changes, prior to implementation.
Canada (HC):
Verify that the Manufacturer has a process or procedure for identifying a “significant change” to a class III or IV device. Verify that information about “significant changes” is submitted in a medical device license amendment application [CMDR 1, 34].
Japan (MHLW):
Confirm that when the Registered Manufacturing Site plans to make a significant change to a manufacturing processes (e.g. sterilization site change, manufacturing site change), the Registered Manufacturing Site notifies the Marketing Authorization Holder so as the Marketing Authorization Holder can take appropriate regulatory actions [MHLW MO169: 29].
4.1.5 Quality Agreements with outsource supplier (document)
 Australia (TGA):
Australia (TGA):
The conditions of marketing authorization (ARTG inclusion) require that Australian Sponsors undertake some regulatory activities including; customer complaint handling (Act s 41FN, Reg 5.8), the management and communication of technical files /technical documentation (Act s 41FN(3)), adverse event reporting (Act s 41FN, Reg 5.7), conducting recalls (Part 4-9), ensuring that the name and address of the Sponsor is provided with the device (Reg 10.2), the storage of devices (Act s 41FN,Reg 5.9), the keeping of complaint and distribution records (Act s 41FN,Reg 5.10), annual reporting for an initial period of three years, of specified information to the TGA (ACT s41FN, Reg 5.11) for Class AIMD, Class III and Class IIb devices that are implantable, information about supply of specified IVDs (Act s41FN, Reg 5.12), ensuring that some devices that would contravene Part 2 of the Poisons standards are not supplied.
Some Sponsors also provide services for the installation and servicing of a device on behalf of the Manufacturer and consequently are to be treated as a supplier to the Manufacturer.
Where a regulatory requirement for a Sponsor intersects with a regulatory requirement or a requirement of ISO13485 for the Manufacturer, the activity is to be treated as an outsourced activity and documented in the Manufacturer’s QMS. See also Task 5 Purchasing.
The requirement of Regulation 10.2 for “ensuring that the name and address of the Sponsor is provided with the device in such a way that the user of the device can readily identify the Sponsor” is only an obligation on the Sponsor. This activity does not need to be included in the Manufacturer’s QMS documentation however the arrangements for the provision of this information should be disclosed in the written agreement between the Manufacturer and the Sponsor. In cases where an activity performed by the Sponsor also includes the provision of information required by Essential Principle 13 (Labels and IFU), or 13A (patient implant cards and leaflets), the Manufacturer must treat the Sponsor as supplier for that activity.
The Sponsor does not need to be treated as a supplier if the scope of the Manufacturer’s quality management system includes the site and activities of the Sponsor. The oversight of activities that are required by legislation to be conducted by the Sponsors are to be clearly documented in the QMS and included in plans for internal audit.
Canada (HC):
Verify that the roles and responsibilities of any regulatory correspondents, importers, distributors, or providers of a service are clearly documented in the medical device organization’s quality management system and are qualified as suppliers and controlled, as appropriate.
4.1.6 QMS software computer validation (SOP +records)
 Free Template here!
Free Template here!
4.2.1 Quality Manual

4.2.1 Quality Policy (document)

4.2.1 Quality Objectives(document)

4.2.3 Medical Device File (FILE)
Some companies use this clause to accommodate US FDA Device Master Record and CE Technical Documentation requirements
4.2.4 Control of documents (SOP)
 Free template here!
Free template here!
Retention time of technical documentation for a minimum 10 years for all device except implants (15 Years for Implants) after the last product has been placed on the market. MDR reference: Article 10(8)
Australia (TGA):
Confirm that Quality Management System documentation and records in relation to a device described in TG(MD)R Sch3 P1 1.9 are retained by the Manufacturer for at least 5 years.
Note that the conditions of marketing authorization (ARTG inclusion) require that Australian sponsors of Class III/AIMD, implantable Class IIb or Class 4 IVDs to keep records of distribution, and records of information relating to any malfunction or deterioration in the characteristics or performance of a device, or any inadequacy in the design, manufacture, labelling, instructions for use or advertising materials of a device, or any use in accordance with, or contrary to, the use intended by the manufacturer of a device, that has led to any complaint or problem in relation to the device, for a period of up to 10 years. (Reg 5.10)
These requirements should be reflected in the written agreement between the Australian Sponsor and Manufacturer and may also be identified in the Manufacturer’s procedures.
Brazil (ANVISA):
Verify that change records include a description of the change, identification of the affected documents, the signature of the approving individual(s), the approval date, and when the change becomes effective [RDC ANVISA 16/2013: 3.1.5]. Confirm that the manufacturer maintains a master list of the approved and effective documents [RDC ANVISA 16/2013: 3.1.5].
Verify that electronic records and documents have backups [RDC ANVISA 16/2013: 3.1.6].
Japan (MHLW):
Confirm that Quality Management System documentation and records in relation to a device are retained for the following periods (5 years for training records and documentation). [MHLW MO169: 8.4, 9.3, 67, 68]. (1) 15 years for ‘specially designated maintenance control required medical devices’ [or one year plus the shelf life for products when the shelf life or the expiry date (hereinafter simply referred to as the “shelf life”) plus one year exceeds 15 years]. (2) 5 years for the products other than the ‘specially designated maintenance control required medical devices’ (or one year plus the shelf life for the products of which the shelf life plus one year exceeds 5 years).
Note: The ‘specially designated maintenance control required medical device’ is defined as below in PMD Act 2.8:
A medical device designated by the Minister of Health, Labour and Welfare after hearing the opinion of the Pharmaceutical Affairs and Food Sanitation Council as those whose potential risk to the diagnosis, treatment or prevention of disease is significant without proper control since this kind of equipment requires expert knowledge and skill in examination for maintenance and inspection, repair and other management.
United States (FDA):
Verify that electronic records and documents have backups [21 CFR 820.180].
4.2.5 Control of records (SOP)
 Retention time of technical documentation for a minimum 10 years for all device except implants (15 Years for Implants) after the last product has been placed on the market. MDR reference: Article 10(8)
Retention time of technical documentation for a minimum 10 years for all device except implants (15 Years for Implants) after the last product has been placed on the market. MDR reference: Article 10(8)
5.5.1 Responsibility and authority (document)
 Identification of personnel with authority to sign the DOC and appointment of PRRC as per MDR reference: Annex IX,2.2(b), first ‘-‘ and article 15
Identification of personnel with authority to sign the DOC and appointment of PRRC as per MDR reference: Annex IX,2.2(b), first ‘-‘ and article 15
5.6.1 Management review (SOP + record)
 Free template here!
Free template here!
6.2 Personnel competence/training (SOP + record)
 Free SOP here!
Free SOP here!
PRRC qualifications should be met and recorded as per MDR reference: Article 15
Personnel with responsibilities with regard to the MDR shall met the competency requirements. This includes persons involved in Clinical Investigation, clinical evaluation, PMS, risk management, QA and RA personnel and Internal auditors as per MDR reference: Article 10(9d)
Brazil (ANVISA):
Confirm that the manufacturer ensures that any consultant who gives advice regarding design, purchasing, manufacturing, packaging, labeling, storage, installation, or servicing of medical devices has proper qualification to perform such tasks. Those consultants shall be contracted as a formal service supplier, according to purchasing controls defined by the manufacturer [RDC ANVISA 16/2013: 2.3.3].
6.3 Requirements for Infrastructure (document + record)

Brazil (ANVISA):
Verify that manufacturing facilities are configured in order to provide adequate means for people flow [RDC ANVISA 16/2013: 5.1.2].
6.3 Requirements for maintenance (document)
 Free template here!
Free template here!
6.4.1 Requirements for work environment (document)
Brazil (ANVISA):
Verify that biosafety standards are used, when applicable [RDC ANVISA 16/2013: 5.1.3.6].
6.4.1 Work Environment monitoring and control (SOP)
Free template here!
6.4.1 Requirements for health, cleanliness and clothing of personnel (document)

6.4.2 Arrangements for the control of contaminated or potentially contaminated product
7.1 Process for Risk Management (document + record)
 Free template here!
Free template here!
Brazil (ANVISA):
Verify that the manufacturer has established and maintains a continuous process of risk management which covers the entire life cycle of the product. Possible hazards must be identified in both normal and fault conditions, including those arising from human factors issues. The risk associated with those hazards, shall be calculated. Risks must be analyzed, evaluated and controlled, as necessary. Effectiveness of risk controls implemented shall be evaluated [RDC ANVISA 56/2001, RDC ANVISA 16/2013: 2.4].
United States (FDA):
Confirm that the manufacturer has identified the possible hazards associated with the device in both normal and fault conditions. The risks associated with the hazards, including those resulting from user error, should be calculated in both normal and fault conditions. If any risk is judged to be unacceptable, it should be reduced to acceptable levels by the appropriate means. Ensure changes to the device to eliminate or minimize hazards do not introduce new hazards [21 CFR 820.30(g); preamble comment 83].
7.2 Customer related process (document + record)
 Importer and distributor agreement to be drawn up in accordance to MDR reference: Article 13 and 14
Importer and distributor agreement to be drawn up in accordance to MDR reference: Article 13 and 14
7.2.3 Arrangements for communicating with customers (document)
 Free template here!
Free template here!
7.3.1 Design and development (SOP + record)
 Free Design Control template here!
Free Design Control template here!
Free Verification and Validation template here!
Process for “Strategy for regulatory compliance” in accordance with MDR reference: Annex IX,2.2(c) 1st “-“
Process for identification of applicable general safety and performance requirements and identification of solutions to fulfil those requirements – taking applicable Common Specifications and, where opted for, harmonized standards or other adequate solutions into account in accordance with MDR reference: Annex IX,2.2(c) 2nd “-“
Process for the clinical evaluation in accordance with MDR reference: Annex IX,2.2(c) 4th “-“
Process for solutions for fulfilling the applicable specific requirements regarding design and construction, including appropriate pre-clinical evaluation, in particular the requirements of Chapter II of Annex I, in accordance with MDR reference: Annex IX,2.2(c) 5th “-“
Process for solutions for fulfilling the applicable specific requirements regarding the information to be supplied with the device, in particular the requirements of Chapter III of Annex I, in accordance with MDR reference: Annex IX,2.2(c) 6th “-“. For implant card refer to MDR reference: Article 18
United States (FDA):
Verify that the design input procedures contain a mechanism for addressing incomplete, ambiguous, or conflicting requirements [21 CFR 820.30(c)].
7.3.2 Design and development planning
Australia (TGA):
Verify that effective planning for design and development is documented, typically as part of a Quality Plan [TG(MD)R Sch3 P1 Cl 1.4(4)].
Canada (HC):
Verify that Manufacturers of Class IV devices maintain a quality plan that sets out the specific quality practices, resources, and sequence of activities relevant to the device [CMDR 32].
7.3.3 Design and development inputs
Australia (TGA):
Verify that the Manufacturer has identified the relevant Essential Principles that apply to the medical device [TG(MD)R Sch1 Essential Principles].
Verify that Manufacturer has taken into account post-production feedback as an input to monitoring and maintaining product requirements and improving product realization processes.
United States (FDA):
For the selected device(s), verify that the medical device organization has the appropriate marketing clearance [510(k)] or pre-market approval (PMA) if distributing the devices in the United States [21 CFR 807].
Australia (TGA):
Confirm that design inputs include the relevant Essential Principles [TG(MD)R – Schedule 1].
Solutions adopted by the Manufacturer for the design and construction of a medical device are to conform to safety principles that are derived from the generally acknowledged state of the art. [TG(MD)R – Sch 1 – EP2] Safety principles are usually identified in internationally recognized standards.
Compliance with any given standard is not mandatory under Australian legislation however it is one way to demonstrate compliance with the Essential Principles.
The TGA is to presume compliance with the relevant Essential Principles if the Manufacturer has applied, in full, a relevant standard that is identified in a Medical Device Standards Order. (See TGA website – For example, ISO 10993). If relevant standards have not been identified as design inputs, ensure that the Manufacturer has documented a rationale to explain why alternatives have been applied to demonstrate compliance with the Essential Principles [TG(MD)R Sch3 Part 1.4(5)(c)(iii)(C)].
7.3.4 Design and development outputs
Australia (TGA):
If relevant standards have not been applied, or not been applied in full, ensure that the Manufacturer has documented a rationale to explain why alternative methods have been applied to demonstrate compliance with the Essential Principles [TG(MD)R Sch3 Part 1.4(5)(c)(iii)(C)].
For devices incorporating a medicinal substance, verify that documentation also identifies the data to be derived from tests conducted in relation to the substance, and its interaction with the device [TG(MD)R Sch 3 Part 1.4(5)(c)(v)].
7.3.4 Design and development validation
Australia (TGA):
For devices contain temperature sensitive material, environmental conditions should be considered for country specific requirement. For example, test samples should be conditioned as per ISTA 2A to cover Australian climate zone (extreme temperature range -29C-50C) for packaging validation.
Australia (TGA):
Verify that records of the validation include clinical evidence as required by the clinical
evidence procedures [TG(MD) Sch3 P1 Cl 1.4(5)(c)(vii) and TG(MD) Sch3 P8].
For more information about the sources and types of clinical evidence and how they may be used to demonstrate compliance with the Australian EPs, auditors may refer to the clinical evidence
guidelines (medical devices)
7.35 Design and development review
United States (FDA):
Verify that procedures ensure that participants include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed [21 CFR 820.30(e)].
7.3.8 Design and development transfer (SOP + record)
Brazil (ANVISA):
Confirm that the manufacture ensures that the design is not released for production until its approval by the persons assigned by the manufacturer and that the person/s assigned review all records required to the design history file in order to ensure it is complete and the final design is compatible with the approved plans, prior to its release. Confirm that this release, including date and manual or electronic signature of the responsible is documented [RDC ANVISA 16/2013: 4.1.9, 4.1.11].
7.3.9 Design and development changes (SOP + record)
Free template here!
Process for substantial change and the need to report to Notified Body in accordance to MDR reference Annex IX 2.4
Australia (TGA):
Verify that the Manufacturer has a process or procedure for notifying the Auditing Organization of a substantial change to the design process or the range of products to be manufactured [TG(MD)R Sch3 Cl1.5].
Verify that the Manufacturer has a process or procedure for identifying a proposed substantial change to the design, or the intended performance, of a Class AIMD or Class III device, and to notify the assessment body prior to implementing the change [TG(MD)R Sch3 P1 Cl 1.6(4)].
If the Manufacturer is also a holder of a TGA Conformity Assessment Certificate, then the Manufacturer is also required to notify the TGA of these changes.
Verify that Manufacturer has taken into account post-production feedback as an input to monitoring and maintaining product requirements and improving product realization processes.
Brazil (ANVISA):
If the medical device evaluated is already registered/notified with ANVISA, verify that the design change was correctly and promptly submitted to ANVISA for approval, when applicable [Brazilian Law 6360/76 – Art. 13].
Canada (HC):
Verify that the manufacturer has a process or procedure for identifying a “significant change” to a Class III or IV medical device. Verify that information about “significant changes” is submitted in a medical device license amendment application [CMDR 1, 34].
Japan (MHLW):
For the Marketing Authorization Holder, confirm if the Marketing Authorization Holder has submitted a new application, a change application, or a change notification to PMDA/ a Registered Certification Body, when applicable [PMD Act 23-2-5.1, 23-2-5.11, 23-2-5.12, 23-2-23.1, 23-2-23.6, 23-2-23.7].
For the Registered Manufacturing Site, confirm if the site has a mechanism to communicate with the Marketing Authorization Holder about device modifications, so the Marketing Authorization Holder can take appropriate actions. If a critical medical device modification has happened in the Registered Manufacturing Site, confirm if the Registered Manufacturing Site has communicated with Marketing Authorization Holder about the change [MHLW MO169: 29].
United States (FDA):
Verify that the medical device organization obtained a new 510(k) or supplement to the pre-market approval if required [21 CFR 807].
7.3.10 Design History File (FILE)
Australia (TGA):
When a Manufacturer applies TG(MD)R Division 3.2 and selects the Full Quality Assurance conformity assessment procedures [TG(MR)R Schedule 3, Part1, (excluding or including clause 1.6)], quality management system procedures for design and development must be available.
In addition, for all classes of devices, the guidance provided for the audit of technical documentation in Annex 1 is to be followed to ensure the availability of objective evidence that demonstrates compliance with the Essential Principles of Safety and Performance.
Brazil (ANVISA):
According to Brazilian legislations, there is no exception to design control.
If design activities are outsourced, verify that the manufacturer has a complete device master record for the device and records of the design transfer to production [RDC ANVISA 16/2013: 4.1.7, 4.2].
Canada (HC):
With respect to Class II devices that are not subject to Design and Development controls, verify that the manufacturer has objective evidence to establish that Class II devices meet the safety and effectiveness requirements of section 10 to 20 [CMDR 9, 10 to 20].
Japan (MHLW):
Class 1 devices are not required to comply with the requirements of MHLW MO169:30-36, which are equivalent to the requirement of design and development in ISO13485 [MHLW MO169:4.1].
7.4.1 Purchasing process (SOP + record)
Free template here!
For critical supplier, a supply agreement in accordance to CE requirements need to be in place which includes unannounced audit from Manufacturer’s Notified Body.
Australia (TGA):
The conformity assessment procedures require that a Manufacturer demonstrates compliance with the Essential Principles through application of a QMS. Hence a Manufacturer must show how risk management principles have been applied during design and construction, including purchasing, to mitigate risk. (EP 2 – TG(MD)R Sch1 P1 2).
The conditions of marketing authorization (ARTG inclusion) require that Australian Sponsors undertake some regulatory activities including; customer complaint handling (Act s 41FN, Reg 5.8), the management and communication of technical files /technical documentation (Act s 41FN(3)), adverse event reporting (Act s 41FN, Reg 5.7), conducting recalls (Part 4-9), ensuring that the name and address of the Sponsor is provided with the device (Reg 10.2), the storage of devices (Act s 41FN,Reg 5.9) and the keeping of complaint and distribution records (Act s 41FN,Reg 5.10). Some Sponsors also provide services for the installation and servicing of a device on behalf of the Manufacturer. Where a regulatory requirement for a Sponsor intersects with a regulatory requirement or a requirement of ISO13485 for the Manufacturer, the activity is to be treated as an outsourced activity and documented in the Manufacturer’s QMS. Verify that the Manufacturer has adequate supplier controls to mitigate risk and ensure the Sponsor fulfils the outsourced activities included in a written agreement [TG Act 41FN] for those activities.
Other activities that may be outsourced to the Sponsor include making applications on behalf of the Manufacturer to the TGA [TG Act s41EB], representing the Manufacturer in interactions with the TGA [TG Act s41FN(3)], as an intermediary in recalls of products [TG(MD)R Sch 3 – Part 1:1.4(3)], in the notification of substantial changes to a kind of medical device (TG Act s41BE) that may require a variation to an entry in the Australian Register of Therapeutic Goods (TG Act s9D), for the provision of records [TG(MD)R Schedule 3 – Part 1:1.5, 1.9 ], or other matters that may be required to allow the Sponsor to fulfill market authorization conditions [TG Act Part 4-5 Div 2].
The requirement of Regulation 10.2 for “ensuring that the name and address of the Sponsor is provided with the device in such a way that the user of the device can readily identify the Sponsor” is only an obligation on the Sponsor. This activity does not need to be included in the Manufacturer’s QMS documentation however the arrangements for the provision of this information should be disclosed in the written agreement between the Manufacturer and the Sponsor. In cases where an activity performed by the Sponsor also includes the provision of information required by Essential Principle 13 (Labels and IFU) or 13A (implant cards and leaflets) the Manufacturer must treat the Sponsor as supplier for that activity.
The Sponsor does not need to be treated as a supplier if the scope of the Manufacturer’s quality management system includes the site and activities of the Sponsor. The oversight of the Sponsors activities should be clearly documented in the QMS and be included in plans for internal audit.
Canada (HC):
Verify that any regulatory correspondent used by the Manufacturer is treated as a supplier and is adequately qualified.
Japan (MHLW):
Marketing Authorization Holder
If the Marketing Authorization Holder (MAH) has outsourced any process that affects product conformity with requirements, to a Registered Manufacturing Site(RMS), then verify the MAH has performed the necessary verification that the RMS has an appropriate quality management system. If the site of a supplier is a Registered Manufacturing Site, then verify the MAH has performed the necessary verification that the supplier has an appropriate quality management system [MHLW MO169: 65].
Registered Manufacturing Site
If the RMS has outsourced any process that affects product conformity with requirements to another RMS, then verify the outsourcing RMS has performed the necessary verification that the outsourced RMS has an appropriate quality management system. If the site of a supplier is a RMS, then verify the purchase controlling RMS has performed the necessary verification that the supplier has an appropriate quality management system [MHLW MO169: 65].
7.4.2 Purchasing information
Brazil (ANVISA):
Confirm that purchase orders are approved by a designated person. This approval, including date and signature, shall be documented [RDC ANVISA 16/2013: 2.5.4].
7.4.3 Verification of purchased product (record)
 Free template here!
Free template here!
Brazil (ANVISA):
Verify that the manufacturer has established and maintains procedures to ensure the retention of components, raw materials, in-process products and returned products until inspections, tests or other specified verifications have been performed and documented [RDC ANVISA 16/2013: 5.3.3].
7.5.1 Production control (SOP + record)
 Free template here!
Free template here!
Australia (TGA):
See Annex 1
Brazil (ANVISA):
Verify that the device history record of the product includes or refers to the following information: date of manufacture; components used; quantity manufactured; results of inspections and tests; parameters of special processes; quantity released for distribution; labeling; identification of the serial number or batch of production; and final release of the product [RDC ANVISA 16/2013: 3.2.1].
Verify that labeling has not been released for storage or use until a designated individual has examined the labeling for accuracy. The approval, including the date, name, and physical or electronic signature of the person responsible, must be documented in the device history record [RDC ANVISA 16/2013: 5.2.2.3].
United States (FDA):
Verify that labeling is not released for storage or use until a designated individual has examined the labeling for accuracy including, where applicable, the correct unique device identifier (UDI) or Universal Product Code (UPC), expiration date, control number, storage instructions, handling instructions, and any additional processing instructions [21 CFR 820.120(b)].
Confirm that labeling is stored in a manner that provides proper identification and prevents mix-ups. Verify labeling and packaging operations are controlled to prevent labeling mix-ups [21 CFR 820.120(c) and (d)].
Verify that the label and labeling used for each production unit, lot, or batch are documented in the batch record, as well as any control numbers used [21 CFR 820.120(e), 820.184(e)].
7.5.2 Requirements for cleanliness of product (document)
Brazil (ANVISA):
Confirm that a pest control program has been established and where chemicals are used as part of the pest control program, the company must ensure that they do not affect product quality [RDC ANVISA 16/2013: 5.1.3.4].
Verify that the manufacturer has established and maintains housekeeping procedures and schedules for production areas and warehouses, in conformance with production specifications [RDC ANVISA 16/2013: 5.1.3.1].
7.5.3 Requirements for medical device installation and acceptance criteria for verification of installation (document + record)

7.5.4 Servicing activities (SOP + record)

Brazil (ANVISA):
Confirm that the manufacturer has established and maintains procedures to ensure that records of servicing activities are kept with the following information:
– the product serviced
– the control number of the product serviced
– the date of completion of service
– identification of the service provider
– description of service performed
– results of inspections and tests performed [RDC ANVISA 16/2013: 8.2.1].
Verify that the manufacturer periodically reviews the records of servicing activities. In cases where the analysis identifies trends that pose danger or records involving death or serious injury, a corrective or preventive action must be initiated [RDC ANVISA 16/2013: 8.2.2].
United States (FDA):
Verify that each manufacturer who receives a service report that represents an event that must be reported to FDA as a medical device report automatically considers the report a complaint [21 CFR 820.200(c)].
Confirm that service reports are documented and include the name of the device serviced, any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used; and the date of service [21 CFR 820.200(d)].
7.5.5 Particular requirements for sterile medical devices (record)

7.5.6 Production/service provision process validation (SOP + record)
 Free template here!
Free template here!
Brazil (ANVISA):
Verify that analytical methods, supporting auxiliary systems for production and environmental control that can adversely affect product quality or the quality system are validated, periodically reviewed and, when necessary, revalidated according to documented procedures [RDC ANVISA 16/2013: 5.5.2, 5.5.3].
United States (FDA):
Process validation is required for sterilization, aseptic processing, injection molding, and welding [21 CFR 820.75; preamble comment 143].
Australia (TGA):
Confirm that methods of validation have regard to the generally acknowledged state of the art (e.g. current Medical Device Standard Orders – MDSO, ISO/IEC Standards, BP, EP, USP etc.) [TG Act s41CB, TG(MD)R Sch 1 P1 2(1)].
7.5.6 Production/service provision software validation (SOP + record)
Free template here!
7.5.7 Validation of processes for sterilization (SOP + record)
Australia (TGA):
Verify that methods of sterilization validation have regard to the generally acknowledged state of the art (e.g. current Australian Medical Device Standard Orders – MDSO, ISO 11135, ISO 11137, Medical Device Standards Order (Endotoxin Requirements for Medical Devices) 2018) [TG(MD)R Sch1 P1 2(1)].
7.5.7 Validation of processes for sterile barrier systems (SOP + record)
7.5.8 Identification (SOP)
 Free template here!
Free template here!
Australia (TGA):
Verify that the design and location of information to be provided with a medical device, including labelling and instructions for use, comply with Essential Principle 13 and implant cards and leaflets with Essential principle 13A.
Brazil (ANVISA):
Verify that the manufacturer has established and maintains procedures to ensure integrity and to prevent accidental mixing of labels, instructions, and packaging materials [RDC ANVISA 16/2013: 5.2.2.1].
Confirm that the manufacturer has ensured that labels are designed, printed and, where applicable, applied so that they remain legible and attached to the product during processing, storage, handling and use [RDC ANVISA 16/2013: 5.2.2.2].
Canada (HC):
Verify that the Manufacturer maintains objective evidence that devices meet the safety and effectiveness requirements. [CMDR 9(2)].
Verify that devices sold in Canada have labeling that conforms to Canadian English and French language requirements and contains the Manufacturer’s name and address, device identifier, control number (for Class III and IV devices), contents of packaging, sterility, expiry, intended use, directions for use and any special storage conditions [CMDR 21-23]. Verify that the Manufacturer maintains distribution records in respect of a device that will permit a complete and rapid withdrawal of the device from the market [CMDR 52-56].
United States (FDA):
If a control number is required for traceability, confirm that a control number is on, or accompanies the device throughout distribution [21 CFR 820.120(e)].
7.5.8 Returned Device Identification (SOP)
7.5.9.1 Traceability (SOP + record)
Free template here!
Brazil (ANVISA):
Verify that the manufacturer maintains distribution records which include or make reference to: the name and address of the consignee, the identification and quantity of products shipped, the date of dispatch, and any numerical control used for traceability [ANVISA RDC 6.3].
Canada (HC):
Verify that the Manufacturer maintains distribution records that contain sufficient information to permit complete and rapid withdrawal of the medical device from the market [CMDR 52-53].
Verify that distribution records of a device are retained by the Manufacturer in a manner that will allow for timely retrieval, for the longer of (a) the projected useful life of the device; and (b) two years after the date the device was shipped [CMDR 55-56].
United States (FDA):
Verify that the Manufacturer maintains distribution records which include or refer to the location of the name and address of the initial consignee, the identification and quantity of devices shipped; and any control numbers used [21 CFR 820.160(b)].
7.5.9.2 Particular requirements for implantable medical devices (record)
Canada (HC):
Verify that the Manufacturer has identified Schedule 2 implants and provides implant registration cards with devices or employs another suitable system approved by Health Canada [CMDR 66-68].
Verify that the Manufacturer of devices that are listed on Schedule 2 of the Medical Devices Regulations maintains distribution records of these devices as well as any information received on implant registration cards related to these Schedule 2 devices [CMDR 54].
United States (FDA):
Verify that the manufacturer has implemented a tracking system for devices for which the manufacturer has received a tracking order from FDA. The tracking system must ensure the manufacturer is able to track the device to the end-user. The manufacturer must conduct periodic audits of the tracking system [21 CFR 821].
7.5.10 Customer property (record)

7.5.11 Preservation of product (SOP + record)
 Free template here!
Free template here!
7.6 Monitoring and measuring equipment (SOP + records)
 Free calibration template here!
Free calibration template here!
Free software validation template here!
8.2.1 Customer feedback system/process (SOP)
 free template here!
free template here!
Post Market Surveillance (PMS) requirement in accordance to MDR reference Article 10(9i) & 10(10)
Post Market Clinical Follow-up (PMCF) in accordance to MDR reference 10 (3)
Australia (TGA):
Verify that the medical device organization has procedures for a post-marketing system that includes a systematic review of post-production experience (e.g. from; expert user groups, customer surveys, customer complaints and warranty claims, service and repair information, literature reviews, post-production clinical trials, user feedback other than complaints, device tracking and registration schemes, user reactions during training, adverse event reports). Investigation should take place in a timely manner to ensure that reporting timeframes for adverse events or the implementation of advisory notices (recalls) may be met by the Australian Sponsor [TG(MD)R Sch3 P1 1.4(3)(a)].
Note: In Australia the conduct of a recall is the responsibility of the Australian Sponsor in accordance with the Australian Uniform Recall Procedure for Therapeutic Goods.
Japan (MHLW/PMDA):
Confirm that the person operating the Registered Manufacturing Site has determined and implemented effective arrangement for communicating with the Japanese Marketing Authorization Holder in relation to customer feedback, including customer complaints, and advisory notices [No.169: 29].
8.2.2 Complaint handling (SOP + record)

Brazil (ANVISA):
Verify that each manufacturer has established and maintains procedures to receive, examine, evaluate, investigate and document complaints. Such procedures must ensure that:
– Complaints are received, documented, analyzed, evaluated, investigated and documented by a formally designated unit
– Where applicable, complaints must be reported to the competent health authority
– Complaints must be examined to determine whether an investigation is necessary. When an investigation is not done, the unit must maintain a record that includes the reason that the investigation was not performed and the name of the persons responsible for the decision.
– Each manufacturer must examine, evaluate and investigate all complaints involving possible nonconformities of the product. Any claim for death, injury or threat to public health must be immediately reviewed, evaluated and investigated.
– The records of the investigation must include:
– Product name
– Date of receipt of the complaint
– Any control number used
– Name, address and telephone number of the complainant
– Nature of complaint
– Data and research results including actions taken [RDC ANVISA 16/2013: 7.2].
Canada (HC):
Verify that the Manufacturer maintains records of reported problems related to the performance characteristics or safety of a device, including any consumer complaints received by the Manufacturer after the device was first sold in Canada, and all actions taken by the Manufacturer in response to the problems referred to in the complaints [CMDR Section 57].
Verify that the Manufacturer has established and implemented documented procedures that will enable it to carry out an effective and timely investigation of the problem reports through the customer complaints, and to carry out an effective and timely recall of the device [CMDR Section 58].
United States (FDA):
Verify procedures have been defined, documented, and implemented for receiving, reviewing, and evaluating complaints by a formally designated unit. Procedures must ensure that:
– All complaints are processed in a uniform and timely manner
– Oral complaints are documented upon receipt
– Complaints are evaluated to determine whether the complaint represents an event which is required to be reported to FDA
Each manufacturer must review and evaluate all complaints to determine whether an investigation is necessary. When no investigation is made, the manufacturer must maintain a record that includes the reason no investigation was made and the name of the individual responsible for the decision not to investigate.
Any complaint of the failure of the device, labeling, or packaging to meet any of its specifications must be reviewed, evaluated, and investigated, unless such investigation has already been made for a similar complaint and another investigation is not necessary.
Any complaint that represents an event which must be reported to FDA must be promptly reviewed, evaluated, and investigated by a designated individual(s) and must be maintained in a separate portion of the complaint files or otherwise clearly identified. Records of investigation must include a determination of:
– Whether the device failed to meet specifications
– Whether the device was being used for treatment or diagnosis
– The relationship, if any, of the device to the reported incident or adverse event
When an investigation is made, a record of the investigation must be maintained by the formally designated unit. The record of investigation must include:
– The name of the device
– The date the complaint was received
– Any unique identifier (UDI), or Universal Product Code (UPC) or any other device identification(s) and control number(s) used
– The name, address, and telephone number of the complainant
– The nature and details of the complaint
– The dates and results of investigation
– Any corrective action taken
When the manufacturer’s formally designated unit is located at a site separate from the manufacturing establishment, the investigated complaint(s) and the record(s) of investigation must be reasonably accessible to the manufacturing establishment [21 CFR 820.198].
8.2.3 Reporting to regulatory authorities (SOP + record)
 Free template for adverse event here!
Free template for adverse event here!
Free template for FSCA here!
Reporting of serious incident, trend reporting and field safety corrective action (FSCA) in accordance to MDR reference 10(9k), 87 – 89
Australia (TGA):
Manufacturers are required to implement a post-marketing system that includes provisions for adverse event reporting – e.g. Therapeutic Goods (Medical Devices) Regulations 2002 Schedule 3 Part 1 Clause 1.4(3)(c)(i). In view of the written agreement between Manufacturers and the Australian Sponsor [TG Act 41FD], events must be reported by the Manufacturer to the TGA, or to the Sponsor, in a timely manner to ensure that a Sponsor can meet their reporting obligations under the Therapeutic Goods (Medical Devices) Regulation 5.7:
– Verify that the Manufacturer or other person becoming aware of an event that represents a serious threat to public health provides information as soon as practicable. The Sponsor is to report the event within 48 hours.
– Verify that the Manufacturer or other person becoming aware of an event that led to the death or serious deterioration in the state of health of a patient, a user, or other person provides information as soon as practicable. The Sponsor is to report the event within 10 days.
– Verify that the manufacturer or other person becoming aware of an event that the recurrence of which might lead to the death or serious deterioration in the state of health of a patient, a user, or other person provides information as soon as practicable. The Sponsor is to report the event within 30 days.
Note: An event that leads to a serious threat to human health is a hazard arising from a systematic failure of the devices or an event or other occurrence that may lead to death or serious injury.
Note: Adverse events may be reported on-line to the TGA, by the Manufacturer or Sponsor, at https://www.tga.gov.au/reporting-problems.
Note: It is a condition on Australian Sponsors of Class AIMD, Class III and Implantable Class IIb devices that they provide three consecutive annual reports to the TGA following inclusion of the device in the ARTG. Annual reports are due 1 October each year. Reports should be for the period 1 July to 30 June. The report is to include:
– ARTG no.
– Product name
– Model no(s)
– Number supplied in Australia
– Number supplied worldwide (Numbers should include devices that are the same but supplied under a different name in another jurisdiction)
– Number of complaints in Australia
– Number of complaints worldwide
– Number of adverse events and incident rates in Australia (Rate= No. of events/ No. Supplied x 100 = Rate%)
– Number of adverse events and incident rates world wide
– A list of the more common complaints and all of the adverse events
– Device Incident Report (DIR) number of those adverse events reported to the TGA
– Regulatory/corrective action/notification by Manufacturer
Note: Australian Sponsors are required to provide Manufacturers with any information that will assist the Manufacturer to comply with the obligations of a conformity assessment procedure (e.g. information in relation to adverse events) [TG(MD)R Reg 5.8].
Brazil (ANVISA):
Verify that a post-market surveillance system is established and implemented in the medical device organization and integrated into the Quality System, with procedures and work flows established to ensure the correct and the prompt identification of adverse events, the performance of investigations and use of the results to improve the safety and effectiveness of the device when necessary [RDC ANVISA 67/2009 – Art. 6º].
For domestic manufacturers (also applies to legal representatives in Brazil) – verify that top management has designated a professional to be responsible for the post-market surveillance system. This designation shall be documented [RDC ANVISA 67/2009 – Art. 5º].
Verify that the medical device organization has mechanisms for processing and recording complaints, conducting investigations, and providing feedback directly to the complainant, or in the case of an international manufacturer, to their legal representative in Brazil, as necessary [RDC ANVISA 67/2009 – Art. 6º, Art. 7º, Art. 9º].
Verify that the medical device organization has notified the regulatory authority about problems associated with their devices, including adverse events (critical or non-critical), any technical defect that was identified regarding products already marketed, anything that can cause a serious hazard to public health, or cases of counterfeit [RDC ANVISA 67/2009 – Art. 8º].
For international manufacturer, verify that the legal representative in Brazil is aware about the occurrence of possibility of death, serious hazard to public health or cases of counterfeit, associated with their products exported to Brazil [RDC ANVISA 67/2009 – Art. 8º].
Canada (HC):
Medical Device Regulations SOR/98-282, Section 59-61.1
Verify that the Manufacturer and the importer of a medical device make a preliminary and final report to the minister concerning any incident occurring inside or outside Canada involving a device sold in Canada:
– Related to the failure of the device or deterioration in its effectiveness or any inadequacy in its labeling or in its directions for use.
– Has led to death or serious deterioration in the state of health of a patient, user, or other person, or could do so if it were to occur [CMDR 59].
Verify that the Manufacturer or other person becoming aware of an event that led to the death or serious deterioration in the state of health of a patient, a user, or other person provides information in a preliminary report within 10 days after the person becomes aware of the event or occurrence [CMDR 60 (1) (a) (i)].
Verify that the Manufacturer or other person becoming aware of an event that the recurrence of which might lead to the death or serious deterioration in the state of health of a patient, a user, or other person provides information in a preliminary report within 30 days after the person becomes aware of the event or occurrence [CMDR 60 (1) (a) (ii)].
Note: The requirement to report incidents meeting the requirements of section 59.(1) that occur outside of Canada does not apply unless the Manufacturer has indicated, to a regulatory agency of the country in which the incident occurred, the Manufacturer’s intention to take corrective action, or unless the regulatory agency has required the Manufacturer to take corrective action. [CMDR 59.(2)]
Verify that Manufacturer has made effective arrangements to submit preliminary reports to the Minister and that the reports contain [CMDR 60 (2)]:
– the identifier of any medical device that is part of a system, test kit, medical device group, medical device family or medical device group family
– if the report is made by:
– the Manufacturer: the name and address of that Manufacturer and of any known importer, and the name, title and telephone and facsimile numbers of a representative of the Manufacturer to contact for any information concerning the incident, or
– the importer of the device: the name and address of the importer and of the Manufacturer, and the name, title and telephone and facsimile numbers of a representative of the importer to contact for any information concerning the incident.
– the date on which the incident came to the attention of the Manufacturer or importer
– the details known in respect of the incident, including the date on which the incident occurred and the consequences for the patient, user or other person
– the name, address and telephone number, if known, of the person who reported the incident to the Manufacturer or importer
– the identity of any other medical devices or accessories involved in the incident, if known
– the Manufacturer’s or importer’s preliminary comments with respect to the incident
– the course of action, including an investigation, that the Manufacturer or importer proposes to follow in respect of the incident and a timetable for carrying out any proposed action and for submitting a final report
– a statement indicating whether a previous report has been made to the Minister with respect to the device and, if so, the date of the report.
If a preliminary report required by section 60 is submitted to the Minister and/or Importer, verify that the Manufacturer has submitted a final report to the Minister in writing in accordance with the timetable established under CMDR 60(2)(h) and the final report contains [CMDR 61(1)]:
– a description of the incident, including the number of persons who have experienced a serious deterioration in the state of their health or who have died
– a detailed explanation of the cause of the incident and a justification for the actions taken in respect of the incident
– any actions taken as a result of the investigation, which may include:
– increased post-market surveillance of the device
– corrective and preventive action respecting the design and manufacture of the device, and recall of the device.
If the reports required by section 60 and 61 are submitted to the Minister just by the Importer, verify that the Manufacturer has advised the Minister in writing that the reports the Manufacturer and importer would have submitted were identical and that the Manufacturer has permitted the importer to prepare and submit reports to the Minister on the Manufacturer’s behalf [CMDR 61.1].
Japan (MHLW):
Marketing Authorization Holders are required to implement post market safety activities in accordance with domestic (Japanese) regulatory requirements in addition to the QMS requirements.
The persons operating the Registered Manufacturing Sites are not required to report any adverse event directly to a Regulatory Authority, but shall report any adverse event which meets the criteria specified by the Ordinance for Enforcement of PMD Act Article 228-20.2 to the Marketing Authorization Holder [MHLW MO169: 62.6].
Verify that the person operating the Registered Manufacturing Site provides events which meets the following criteria defined by the Ordinance for Enforcement of PMD Act Article 228-20.2 (see below), to the Marketing Authorization Holder in a timely manner.
– The following malfunction events which may cause or may have caused health damage:
– Serious event (domestic and foreign)
– Unlabeled non-Serious event (domestic)
– The following Adverse Reaction events which was caused or might have been caused by the malfunction of a medical device:
– Serious event (domestic and foreign)
– Unlabeled non-Serious event (domestic)
– Any action taken for preventing the occurrence or expansion of public health hazard in relation to a medical device which is marketed in foreign countries and is equivalent to the one marketed in Japan. The action includes but not limited to:
– Suspension of manufacturing, importing or selling
– Recall and
– Abolishment.
– Study report that indicates:
– Possibility of event of cancer and other serious illness, injury or death caused by malfunction of a medical device (domestic and foreign), or by infectious disease arising from usage of a device (domestic and foreign)
– Significant occurrence rate change of event etc. caused by malfunction of a medical device (domestic and foreign)
– Significant occurrence rate change of infectious disease caused by usage of a medical device (domestic and foreign)
– The fact that a medical device is less effective than claimed when approved.
United States (FDA):
21 CFR 803: Medical Device Reporting
Determine whether the manufacturer has developed a process for reporting to FDA incidents involving device-related deaths, serious injuries, and reportable malfunctions that occur within and outside the United States if the same or similar device is marketed to the United States.
Confirm that the manufacturer has developed, maintained, and implemented written medical device reporting (MDR) procedures for the following:
– Internal processes that provide for:
– Timely and effective identification, communication, and evaluation of events that may be subject to MDR requirements
– A standardized review process or procedure for determining when an event meets the criteria for reporting
– Timely transmission of complete medical device reports to FDA
– Documentation and recordkeeping requirements for:
– Information that was evaluated to determine if an event was reportable;
– All medical device reports and information submitted to FDA
– Processes that ensure access to information that facilitates timely follow-up and audit.
Verify that reports are made within 30 calendar days after the day that the manufacturer receives or otherwise becomes aware of information, from any source, that reasonably suggests that a device that is marketed may have caused or contributed to a death or serious injury:
– Confirm the manufacturer’s MDR files contain the following:
– Information (or references to information) related to the adverse event, including all documentation of deliberations and decision-making processes used to determine if a device- related death, serious injury, or malfunction was or was not reportable to FDA
– Copies of all MDR forms and other information related to the event submitted to FDA.
If a device has malfunctioned and this device or a similar device that is marketed would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur, quarterly summary reporting is acceptable for most device product codes.
If the manufacturer maintains MDR event files as part of the complaint file, ensure that the manufacturer has prominently identified these records as MDR reportable events. FDA will not consider a submitted MDR report to comply with 21 CFR 803 unless the manufacturer evaluates an event in accordance with the quality management system requirements. Confirm that the manufacturer has documented and maintained in the MDR event files an explanation of why the manufacturer did not submit or could not obtain any information required by 21 CFR 803, as well as the results of the evaluation of each event.
Compare the information submitted on the individual medical device report to the information contained in the associated complaint and confirm the medical device report contains all information related to the event that is reasonably known to the manufacturer.
Verify the manufacturer has submitted reports to FDA no later than 5 work days after the day that the manufacturer becomes aware that:
– An MDR reportable event necessitates remedial action to prevent an unreasonable risk of substantial harm to the public health. The manufacturer may become aware of the need for remedial action from any information, including any trend analysis; or
– FDA has made a written request for the submission of a 5-day report. If the manufacturer receives such a written request from FDA, the manufacturer must submit, without further requests, a 5-day report for all subsequent events of the same nature that involve substantially similar devices for the time period specified in the written request. FDA may extend the time period stated in the original written request if FDA determines it is in the interest of the public health.
Verify the manufacturer submitted supplemental reports within one month of obtaining information that was not submitted in an initial report.
Confirm that medical device reports include the unique device identifier (UDI) that appears on the device label or on the device package.
Medical device reports submitted to FDA must be submitted electronically via the Electronic Submissions Gateway (ESG) using eSubmitter or the AS2 Gateway-to-Gateway using HL7 ICSR XML software.
Australia (TGA):
Manufacturers are required to implement a post-marketing system that includes provisions for the recovery of devices – e.g. Therapeutic Goods (Medical Devices) Regulations 2002 Schedule 3 Part 1 Clause 1.4(3)(c)(ii). In view of the written agreement between Manufacturers and the Australian Sponsor [TG Act 41FD] (see Annex 4) proposed recalls must be reported by the Manufacturer to the TGA, or to the Sponsor in a timely manner to ensure that a Sponsor can meet their reporting obligations [Therapeutic Goods (Medical Devices) Regulation 5.7, Therapeutic Goods Act Part 4-9 and the Uniform Recall Procedure for Therapeutic Goods (URPTG)].
Note: Further information concerning the Australian requirements for advisory notices and the recovery of devices is available at https://www.tga.gov.au/recalls
Note: Australian Sponsors are required to provide Manufacturers with any information that will assist the Manufacturer to comply with the obligations of a conformity assessment procedure (e.g. information in relation to the recovery of devices) [TG(MD)R Reg 5.8].
Brazil (ANVISA):
Verify that procedures and work flows were established in order to identify when field actions (recalls and corrections) are necessary, in accordance with the medical device organization’s post-market surveillance system and quality system [RDC ANVISA 67/2009 – Art. 6º, RDC ANVISA 23/2012 – Art. 1º, Art. 5º].
Verify that the medical device organization keeps records regarding field actions performed, including those that do not need to be reported to regulatory authorities [RDC ANVISA 23/2012 – Art. 4º; Art. 6º, Art. 10, Art. 11, Art. 16].
For domestic manufacturers (also applies to legal representatives in Brazil) – verify that the medical device organization has sent to the regulatory authority the reports requested, according Brazilian regulation [RDC ANVISA 23/2012– Art. 10, Art. 11].
Verify that the medical device organization has performed field actions based on potential or concrete evidence that their product does not comply with essential requirements of safety and effectiveness [RDC ANVISA 23/2012 – Art. 4º, Art. 6º, Art. 7º, Art. 13, Art. 14, Art. 15].
For domestic manufacturers (also applies to legal representatives in Brazil) – verify that the medical device organization has performed field actions when required by the regulatory authority [RDC ANVISA 23/2012 – Art. 6º].
For domestic manufacturers (also applies to legal representatives in Brazil) – verify that the medical device organization notified the regulatory authority regarding field actions, in accordance with requirements and deadlines established per Brazilian regulation [RDC ANVISA 23/2012 – Art. 7º, Art. 8º].
For international manufacturers, verify that the legal representative in Brazil was aware about the occurrence of field actions performed on products exported to Brazil [RDC ANVISA 67/2009 – Art. 8º].
Canada (HC):
Medical Device Regulations SOR/98-282, Section 63 – 65.1:
Verify that the Manufacturer and the importer of a medical device, on or before undertaking a recall of a device provide the minister with the following information [CMDR 64]:
– the name of the device and its identifier, including the identifier of any medical device that is part of a system, test kit, medical device group, medical device family or medical device group family
– the name and address of the Manufacturer and importer, and the name and address of the establishment where the device was manufactured, if different from that of the Manufacturer
– the reason for the recall, the nature of the defectiveness or possible defectiveness and the date on and circumstances under which the defectiveness or possible defectiveness was discovered
– an evaluation of the risk associated with the defectiveness or possible defectiveness
– the number of affected units of the device that the Manufacturer or importer:
– manufactured in Canada,
– imported into Canada,
– sold in Canada.
– the period during which the affected units of the device were distributed in Canada by the Manufacturer or importer
– the name of each person to whom the affected device was sold by the Manufacturer or importer and the number of units of the device sold to each person
– a copy of any communication issued with respect to the recall
– the proposed strategy for conducting the recall, including the date for beginning the recall, information as to how and when the Minister will be informed of the progress of the recall and the proposed date for its completion
– the proposed action to prevent a recurrence of the problem
– the name, title and telephone number of the representative of the Manufacturer or importer to contact for any information concerning the recall.
Verify that as soon as possible after the completion of the recall the Manufacturer and the importer reports to the minister the results of the recall and the action taken to prevent a recurrence of the problem [CMDR 65].
If the reports required by section 64 and 65 are submitted to the Minister just by the Importer, verify that the Manufacturer has advised the Minister in writing that the reports the Manufacturer and importer would have submitted were identical and that the Manufacturer has permitted the importer to prepare and submit reports to the Minister on the Manufacturer’s behalf [CMDR 65.1].
Japan (MHLW):
Marketing Authorization Holders are required to report advisory notices to Regulatory Authorities [PMD Act 68-11].
Confirm that the person operating the Registered Manufacturing Site has determined and implemented effective arrangement for communicating with the Marketing Authorization Holder in relation to advisory notices [MHLW MO169: 29].
Note: Persons operating Registered Manufacturing Sites are not required to report any advisory notice directly to regulatory authority, but shall communicate with the Marketing Authorization Holder, so they can take necessary regulatory actions.
United States (FDA):
21 CFR 806: Medical Devices; Reports of Corrections and Removals
Verify that the manufacturer has a process in place to notify FDA in the event of actions concerning device corrections and removals and to maintain records of those corrections and removals.
Verify that the written report to FDA of any correction or removal initiated to reduce a risk to health or remedy a violation of the U.S. Food, Drug and Cosmetic Act is reported within 10 working days of initiating the correction or removal. Confirm that the report contains the unique device identifier (UDI) that appears on the device label or on the device package, or the device identifier, Universal Product Code (UPC), model, catalog, or code number of the device and the manufacturing lot or serial number of the device or other identification number.
Confirm that the manufacturer maintains records of any correction and removal not required to be reported to FDA (e.g. corrections and removals conducted to correct a minor violation of the U.S. Food, Drug and Cosmetic Act or no risk to health). Confirm that records of corrections and removals not required to be reported contain the unique device identifier (UDI) that appears on the device label or on the device package, or the device identifier, Universal Product Code (UPC), model, catalog, or code number of the device and the manufacturing lot or serial number of the device or other identification number.
8.2.4 Internal audits (SOP + record)
 Free template here!
Free template here!
8.2.6 Monitoring and measuring of product (SOP + record)
Brazil (ANVISA):
Verify that sampling plans are defined and based on valid statistical rationale. Each manufacturer must establish and maintain procedures to ensure that sampling methods are suitable for their intended use and are reviewed regularly. A review of sampling plans should consider the occurrence of nonconforming product, quality audit reports, complaints and other indicators [RDC ANVISA 16/2013: 9.2].
United States (FDA):
Verify that the manufacturer establishes and maintains procedures to ensure that sampling methods are adequate for their intended use and ensure that when changes occur, the sampling plans are reviewed [21 CFR 820.250(b)].
8.3.1 Control of nonconforming product (SOP + record)
 Free Template here!
Free Template here!
8.3.2 Actions in response to nonconforming product detected before delivery (record)
8.3.3 Issuing of advisory notices (SOP + records)
Free template here!
8.3.4 Rework (SOP + record)

8.4 Analysis of data (SOP + record)

8.5.2 Corrective action (SOP + record)
 Free template here!
Free template here!
Brazil (ANVISA):
Verify that the manufacturer has ensured that information about quality problems or nonconforming products are properly disseminated to those directly involved in the maintenance of product quality and to prevent occurrence of such problems [RDC ANVISA 16/2013: 7.1.1.6].
United States (FDA):
Verify procedures ensure that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of problems [21 CFR 820.100(a)(6)].
Confirm procedures provide for the submission of relevant information on identified quality problems, as well as corrective and preventive actions, for management review [21 CFR 820.100(a)(7)].
8.5.3 Preventive action (SOP + record)
Free template here!
Contact
Contact us here to learn more about Easy Guide on how to comply to MDR and ISO 13485

